NGS-genome sequencing
Wiki error: Section name cannot be repeated ''
Table of contents
Back to index
NGS [edit]
(Paolo Piovani)
In the 1970s, Sanger and colleagues and Maxam and Gilbert developed methods to sequence DNA by chain termination and fragmentation techniques, respectively. This transformed biology by providing the tools to decipher complete genes and, later, entire genomes. Thanks to these advances, the Sanger technique ultimately enabled the completion of the first human genome sequence in 2004.
The growing number of sequencing projects, such as the Human Genome Project, required vast amounts of time and resources and it was clear that faster, higher throughput, and cheaper technologies were required. This stimulated the development and commercialization of next-generation sequencing (NGS) technologies. These new sequencing methods share three major improvements.
1) Instead of requiring bacterial cloning of DNA fragments they rely on the preparation of NGS libraries in a cell free system.
2) Instead of hundreds, thousands-to-many-millions of sequencing reactions are produced in parallel.
3) The sequencing output is directly detected without the need for electrophoresis; base interrogation is performed cyclically and in parallel. The enormous numbers of reads generated by NGS enabled the sequencing of entire genomes at an unprecedented speed.
The only main drawback of NGS technology was their relatively short reads, which made genome assembly more difficult and required new alignment algorithms.
Pyrosequencing [edit]
(modified by Francesco De Giorgio)
The first NGS technology to be released in 2005 was the pyrosequencing method by 454 Life Sciences (now Roche).
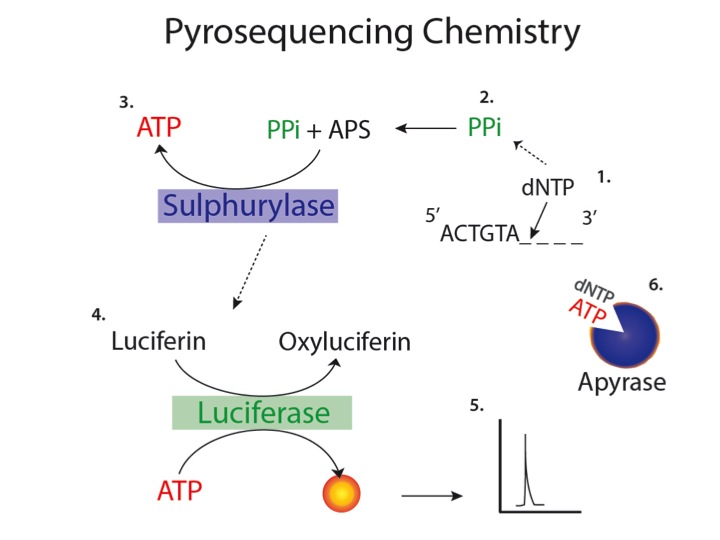
The first event of the pyrosequencing reaction is the incorporation of nucleotide in the new filament by DNA polymerase. One of the four of dNTPs (2’-deoxynuclotides) is added to the reaction. At this point if the nucleotide is incorporated, PPi (pyrophosphate) is released and can be used to produce ATP. The reaction is catalysed by ATP sulphurylase, transferring PPi to the 5’ of APS (5’-phosphosolphoadenosine), generating ATP and sulphate. ATP (that of course cannot be incorporated into the DNA as it carries OH at the 2’) is used by luciferase to convert luciferin into oxyluciferin and producing light, that is detected by a detector. Unincorporated nucleotides and ATP are degraded by the apyrase, and the reaction can restart with another nucleotide. Each peak that is obtained is associated to the incorporation of one nucleotide: if we have multiple peaks during one single step it means that we have the incorporation of the same nucleotide in a number that is equal to the number of peaks.
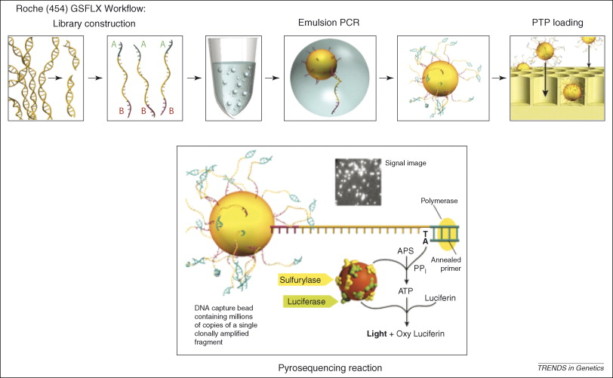
This kind of technology was implemented by Roche 454. Firstly, DNA is randomly fragmented and then, adaptors are ligated both at the 5’ and 3’ of amplicons: in this way fragments are univocally “marked”. DNA is denaturated and hybridized with beads containing adaptors. Hybridization produces beads at which we have one type of molecule (containing the same adaptor). Beads are in suspension in a water solution and are put together with reagents of the PCR reaction and oil. This produce an emulsion leading to have one beads for droplet. For this reason, the following step is called emulsion PCR, leading to have beads containing million of copies of a single clonally amplified fragment. Beads are dropped on specific nano-wells, in which one single bead can drop. Amplification has the role to increase signal coming from the pyrosequencing reaction.
The 454 Genome sequencer generated about 200 000 reads (20 Mb) of 110 bp. Nine years after, it could generate more thatn 700 Mb per run in 10 hours with read length of 1000 nt.
Nowadays, instead of using luciferase, the modern pyrosequencing is based on Ion proton. It based on the realizing of H+, as a result of the formation of the phosphodiester bond. In this case the incorporation of the nucleotide is detected in term of changes in pH in the nano-well. One problem with this approach is related to multiple incorporation of the same nucleotide: in this case there is no change in pH value, so that the signal loses its linearity.

In 2006, the Solexa/Illumina sequencing platform was commercialized. Now, Illumina
has achieved a particularly impressive increase in throughput., which offers
the highest throughput per run and the lowest per-base cost.


The third technology to be released was Sequencing by Oligo Ligation Detection (SOLiD) by Applied Biosystems (now Life Technologies) in 2007.

The Illumina and SOLiD sequencers generated much larger numbers of reads than 454 (30 and 100 million reads, respectively) but the reads produced were only 35 bp long.
In 2010, Ion Torrent (now Life Technologies) released the Personal Genome Machine (PGM). This system was developed by Jonathan Rothberg, the founder of 454, and resembles the 454 system. An important difference is that the PGM uses semiconductor technology and does not rely on the optical detection of incorporated nucleotides using fluorescence and camera scanning. This resulted in higher speed, lower cost, and smaller instrument size. The first PGM generated up to 270 Mb of sequence with up to 100 nt reads; slightly shorter than those produced by 454.
Other NGS methods have been developed, such as Qiagen intelligent bio-systems sequencing-by-synthesis.
Polony sequencing, and a single molecule detection system (Helicos BioSciences).
In this latter system, the template DNA is not amplified before sequencing,
which places this method at the interface between NGS and the so-called third generation
sequencing technologies.
Third-generation methods also allow the detection of single molecules but as an additional common feature sequencing occurs in real time. The leader in this field is currently Pacific Biosciences (PacBio)
Their first instrument, the PacBio RS, appeared in 2010 and generated several thousands of up-to-several-kilobase-long reads. The long reads made this technology ideal for the completion of de novo genome assemblies. PacBio is based on the detection of natural DNA synthesis by a single DNA polymerase. Incorporation of phosphate-labelled nucleotides leads to base-specific fluorescence, which is detected in real time. Sequencing runs therefore last minutes or hours rather than days.
The advent of NGS immediately revolutionized genomics research by bringing the sequencing of entire genomes within reach of many small laboratories. In addition, gene expression studies frequently changed from using microarrays to NGS-based methods, which enabled the identification and quantification of transcripts without prior knowledge of a particular gene and provided information regarding alternative splicing and sequence variation. For genome-wide mapping of protein–DNA interactions and epigenetic marks, chromatin immunoprecipitation followed by sequencing (ChIP-seq) was another early application of NGS. Microarrays had been used before, but ChIP-seq provided substantially improved data with higher resolution and a larger dynamic range.
The advent of NGS has enabled researchers to study biological systems at a level never before possible. As the technologies have evolved, an increasing number of sample preparation methods and data analysis tools have spawned an immense diversity of scientific applications. NGS has thus become a key technology in basic science and is rapidly becoming an established tool in translational research as well. Ongoing cost reduction and the development of standardized pipelines will probably make NGS a standard tool for more-routine applications in the near future. The significant increase in sample throughput would be beneficial in this field, and NGS methods may be able to solve specific cases that would not be possible with Sanger sequencing. An enormous benefit of introducing large-scale sequencing into clinical research and ultimately healthcare is joining genomic and phenotypic information related to health and disease to find new genetic associations. This will advance our understanding of the functional consequences of DNA mutations and improve our ability to diagnose and predict outcomes of diseases for individual patients. Eventually, this should bring personalized medicine closer to a reality. Other promising technologies are starting to appear, however.
An example is Nanopore sequencing, which is based on the transit of a DNA molecule through a pore while the sequence is read out through the effect on an electric current or optical signal. Nanopore is considered a third-generation technology because it enables the sequencing of single molecules in real time. Major advantages are direct sequencing of DNA or RNA molecules without the need for library preparation or sequencing reagents. Very recently the first commercially available sequencer that uses this technology, the MinIONTM, was pre-released by Oxford Nanopore Technologies. Unlike the bulky, expensive sequencing machines on the market, the MinIONTM is an inexpensive hand-held device. It is capable of producing reads of up to 10 kb, but the data are still of insufficient quality owing to systematic errors and further improvement will therefore be required.

These novel technologies clearly need to be improved and whether or not they will shake up the sequencing industry or if Illumina will remain the leading technology in the coming years remains to be seen. However, they have the potential to launch another revolution in DNA sequencing and its applications. For example, they might be able to sequence single RNA or even protein molecules directly, vastly increasing the ease of single cell genomics.
(for further information see: https://doi.org/10.1016/j.tig.2014.07.001)