Transcriptome analysis: special techniques, RNA-seq, GRO-seq, CAGE, etc.
(Restore this version)
Modified: 18 March 2018, 2:54 PM User: Danilo Lombardi →
Transcriptome analysis: RNA-seq
Overview:
RNA-seq is an high throughput technology used to identify the presence and the quantity of RNA in a biological sample in a given moment. It provides far more precise measurement of levels of transcripts and their isoforms than other methods, allowing researchers to better analyze the transcriptome: the the complete set of transcripts in a cell, and their quantity, for a specific developmental stage or physiological/pathological condition. The key aims of transcriptomics are:
- to catalogue all species of transcript, including mRNAs, non-coding RNAs and small RNAs;
- to determine the transcriptional structure of genes, in terms of their start sites, 5′ and 3′ ends, splicing patterns and other post-transcriptional modifications;
- to quantify the changing expression levels of each transcript during development and under different conditions
In general, a population of RNA is converted into a cDNA library with adaptors attached at one or both ends. Then, each molecule is sequenced to obtain informations from one end (single-end sequencing) or both ends (pair-end sequencing). The sequenced reads (generally 30-400 bp long, depending on the used machinery) are then aligned on a reference genome or transcriptome or de novo assembled to produce a genome-scale transcription map (also expression levels of different genes might be reported).

Respect to other technologies used to investigate transcriptome, RNA-seq has different advantages:
- It is not limited to detecting transcripts derived from an existing genomic sequence;
- It can reveal the precise location of transcription boundaries, to a single-base resolution;
- 30-bp short reads give information about how two exons are connected, whereas longer reads or pair-end short reads should reveal connectivity between multiple exons;
- It can be used to identify variations (as SNPs) in the transcribed region.
- It has very low background signal because DNA sequences can been unambiguously mapped to unique regions of the genome;
- It does not have an upper limit for quantification, which correlates with the number of sequences obtained;
- It is highly accurate for quantifying expression levels;
- Finally, because there are no cloning steps, and with the Helicos technology there is no amplification step, it requires less RNA sample.
Considering all this advantages, RNA-Seq is the first sequencing-based method that allows the entire transcriptome to be surveyed in a very high-throughput and quantitative manner.
Library preparation:
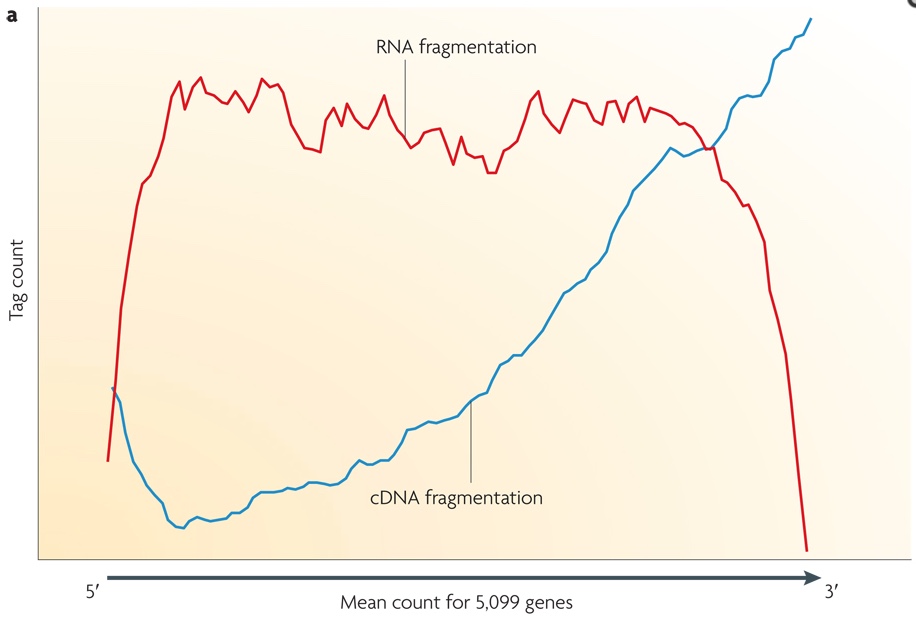
Small RNAs (as miRNAs, siRNAs, etc...) can be directly sequenced after adaption ligation, while long RNAs should be broken into fragments of 200-500 bps to be compatible to the most deep-sequencing technologies. Generally, both RNAs (using RNA hydrolysis or nebulization) and cDNAs (using DNase I treatment or sonication) can be fragmented. Each method has its own bias: RNA fragmentation is depleted for 5' and 3' of each read, while cDNA fragmentation is biased mainly at 3' of the transcript. In the image below, the tag count is the average sequencing coverage.
(Danilo Lombardi)