NGS-genome sequencing
(Restore this version)
Modified: 26 March 2019, 7:42 PM User: Maria Giovanna Caruso →
Return to index
Tentative paragraphs:
(Dallorto Eleonora)
1. Illumina sequencing
Introduction to NGS
Next generation sequencing (NGS), massively parallel or deep sequencing are related terms that describe a DNA sequencing technology . NGS is considered the catch-all term used to describe a number of different modern sequencing technologies. Using NGS an entire human genome can be sequenced within a single day. In contrast, the previous Sanger sequencing technology, used to decipher the human genome, required over a decade to deliver the final draft. The short read, massively parallel sequencing technique has been a fundamentally different approach, which revolutionized sequencing launching the “next generation” in genomic science.
Illumina® sequencing technology was developed by Shankar Balasubramanian and David Klenerman of Cambridge University, later founder of Solexa, a company acquired by Illumina®. (Go for a brief history of Illumina® sequencing https://emea.illumina.com/science/technology/next-generation-sequencing/illumina-sequencing-history.html?langsel=/it/)
Illumina® sequencing is based on the incorporation of reversible dye terminators that enable the identification of single bases as they are incorporated into DNA strands.
This technique offers some advantages over traditional sequencing methods such as Sanger sequencing. The automated nature of Illumina® sequencing makes it possible to sequence multiple strands at once and obtain actual sequencing data quickly. The innovative and flexible sequencing system enables a broad array of applications in genomics, transcriptomics, and epigenomics.
The Basics of NGS Chemistry
A DNA polymerase catalyses the incorporation of fluorescently labelled deoxyribonucleotide triphosphates (dNTPs) into a DNA template strand during sequential cycles of DNA synthesis. During each cycle, at the point of incorporation, the nucleotides are identified by fluorophore excitation. The critical difference is that, instead of sequencing a single DNA fragment, NGS extends this process across millions of fragments in a massively parallel manner.
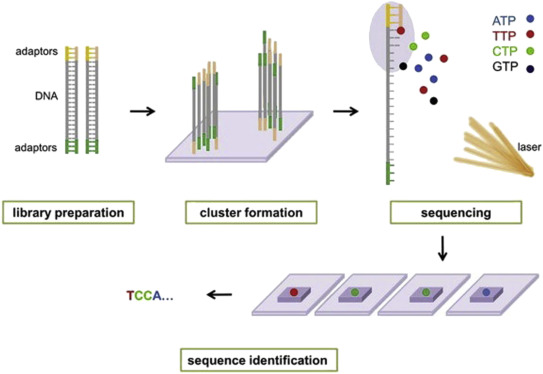
Illumina® sequencing technology works in four basic steps:
- Library Preparation: The first step is to break up the DNA into more manageable fragments of around 200 to 600 base pairs. Short sequences of DNA called adaptors are attached to the DNA fragments. Alternatively, “tagmentation” combines the fragmentation and ligation reactions into a single step: enzymes called transposases randomly cut the DNA into short segments ("tags"), which greatly increases the efficiency of the library preparation process. Adapter-ligated fragments are then PCR amplified and gel purified.
- Cluster Generation: The library is loaded into a flow cell where fragments are captured on a lawn of surface-bound oligos complementary to the library adapters. The complementary DNA binds to primers on the surface of the flowcell and DNA that does not attach is washed away. Unlabeled nucleotide bases and DNA polymerase are then added to lengthen and join the strands of DNA attached to the flowcell. This creates “bridges” of double-stranded DNA between the primers on the flowcell surface. The double-stranded DNA is then broken down into single-stranded DNA using heat, leaving several million dense clusters of identical DNA sequences. When cluster generation is complete, the templates are ready for sequencing.
- Sequencing: Illumina® uses a proprietary reversible terminator based method that detects single bases as they are incorporated into DNA template strands. Primers and fluorescently labeled terminators (terminators are a version of nucleotide base (A, C, G or T) that stop DNA synthesis) are added to the flow cell. The primer attaches to the DNA being sequenced. The DNA polymerase then binds to the primer and adds the first fluorescently labelled terminator to the new DNA strand. Once a base has been added no more bases can be added to the strand of DNA until the terminator base is cut from the DNA. Each of the terminator bases (A, C, G, and T) give off a different colour. The fluorescently labelled terminator group is then removed from the first base and the next fluorescently base can be beside added. And so, the process continues until millions of clusters have been sequenced.
- Data Analysis: During data analysis and alignment, the newly identified sequence reads are aligned to a reference genome, this looks for matches or changes in the sequenced DNA. Following alignment, many variations of analysis are possible: DNA sequencing may be used to determine the sequence of individual genes, larger genetic regions (clusters of genes), full chromosomes, or entire genomes.
For a detailed animation of Illumina® sequencing technology click and watch this video -->
2. Paired-end sequencing
3. Ion Torrent sequencing
4. Roche sequencing
(Maria Giovanna Caruso)
Roche sequencing is another technology of NGS; it’s able to work on genomic DNA, DNA produced by PCR and cDNA.
Sequence length
of 400 – 1000 bases are read, so, at the starting point it is necessary to
reduce the sequence, thanks to a nebulizer, in smaller fragments (fragmentation). After creating single
strand fragments, it’s necessary to bind two different adaptors (for example A
and B) at the two ends (adaptors ligation):
one of the adaptors helps the DNA fragment to attack the beads (insoluble small
particles or nano spheres), since it is complementary to the sequence attacked
to the bead. So, small single strand fragments of DNA will surround the beads. Then, there is the amplification of the
target to get better and accurate sequencing results: to do this, there is
firstly the production of chains starting from the single strand fragments, and
then, these complementary sequences will be used for the amplification (having terminal
sequences complementary to the primers). Now, the beads can be picked and loaded
in specific wells to be inserted in the sequencing machine (loading).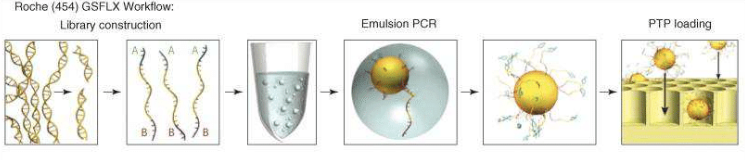
The next step is the pyrosequencing reaction: the main innovation of 454 Roche is the use of inorganic pyrophosphate instead of fluorescent nucleotides, to generate the fluorescence. PPi is the substrate of the enzyme sulfurylase that produce ATP from PPi and AMP. ATP is used by a luciferase that oxidises the luciferin, creating light signals. Nucleotides are added one by one to analyse the fluorescence deriving from the ones that have bound the target sequence.
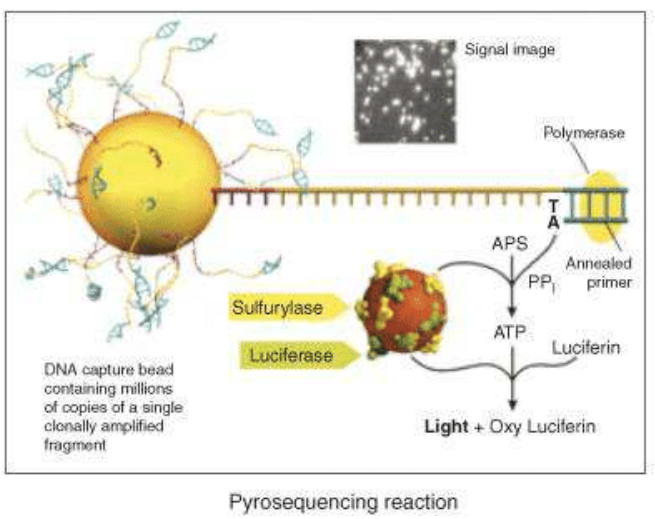
A photodetector can, finally, read the signals and produce a graph expressing the correct sequence. Roche technology currently uses two systems based on the pyrosequencing technology: the GS FLX+ and the GS Junior System. The GS FLX+ System promises read lengths of approximately 1000 base pairs while the GS Junior System promises 400 base pair reads (data analysis).
One of the advantages of 454 systems is their high running speed [sample/day 3840].
There are also some disadvantages:
· the 454 system is prone to errors when estimating the number of bases in a long string of identical nucleotides (homopolymer error);
· the price of reagents is relatively more expensive compared with other next-generation sequencers.
5. Pacific Bio and Nanopore long-run sequencing