NGS
(Restore this version)
Modified: 20 March 2018, 6:17 PM User: Marta Forcella →
Videos are available in the very last section "Auxiliaries".
Subjects to be discussed;
1) When using short-read with high numbers of reads; when using longer-reads with lower numbers?
2) what is Paired-end sequencing ?
3) NGS sequencing of very short RNAs (e.g. micro-RNAs, tRNAs etc.)
4) "exome" sequencing and "Targeted" sequencing
5) How does Pacific Bio long-run sequencing work ?
6) How does Nanopore sequencing work ?
7) other (your proposals)
Next-generation sequencing (NGS) is a massively parallel sequencing technology that has revolutionized the biological sciences. With its ultra-high throughput, scalability, and speed, NGS enables researchers to perform a wide variety of applications and study biological systems at a level never before possible. The most important innovations of NGS regard the (1) lack of DNA/RNA fragment cloning, (2) the use of micro- or nano-reactors immobilized on distinct solid supports, enabling a very high level of parallelization of in situ sequencing and (3) the absence of electrophoretic separation of fragments because once nucleotides are incorporated in the sequencing reaction they are simultaneously identified.
To be analyzed the DNA is fragmented with chemical or enzymatic methods in defined fragments obtaining library of fragments that are subjected to covalent link with adaptators and subsequently used for the clonal amplification and sequencing.
The length of the reads can be different depending on the NGS technology exploited: Illumina’ reads length is around 300, for Abi SOLiD is 75, for 454 is around 1000, exc. Short reads are mainly used for the re-sequencing when mapping against a reference genome, to explore genetic variations as SNVs, indels, CNVs. Platforms with high number of reads and short length are preferable for tag-based RNA sequencing. Whereas longer reads are used for de novo sequencing, starting for primary data lacking a genome of reference. In contrast to short reads, long reads offer the possibility to span repetitive or otherwise difficult regions in the genome, resulting in strongly reduced fragmentation of the assemblies.
Moreover, with long reads, it may be possible to concatenate transcript tags into longer sequences; to characterize transcripts and monitor transcript structures and alternative splicing events longer reads are extremely helpful. Longer reads should, a priori, increase the level of uniquely mapping reads, but such longer reads have an increased cost in reagents and an increase in running time for the instrument.
The paired-end sequencing allows users to sequence both ends of a fragment: after sequencing using a primer for a specific strand of it, the following step uses the opposite primer to sequence the anti-strand. It generates high-quality, alignable sequence data. Moreover, it facilitates detection of genomic rearrangements (as insertions, deletions and inversions) and repetitive sequence elements, as well as gene fusions and novel transcripts. Paired-end DNA sequencing reads provide superior alignment across DNA regions containing repetitive sequences, and produce longer contigs for de novo sequencing by filling gaps in the consensus sequence. The contigs can in turn be ordered to form scaffold on the basis of information of connectivity provided by the pairs of sequences that derive from the ends of the same clone (paired-end). In fact, if the two portions of a paired-end sequence map on two different contigs it is possible that those contigs are contiguous in the genome.(Federica Galvagno)
NGS of short RNA
RNA sequencing (RNA-Seq) uses the capabilities of high-throughput sequencing methods to provide insight into the transcriptome of a cell. Beyond quantifying gene expression, the data generated by RNA-Seq facilitate the discovery of novel transcripts, identification of alternatively spliced genes, and detection of allele-specific expression.
MicroRNAs (miRNAs) are small RNA molecules of 17 to 24 bp that play an important role in the regulation of gene expression by modulating translation and stability of mRNA.
With small RNA-Seq it is possible to discover novel miRNAs and other small noncoding RNAs, and examine the differential expression of all small RNAs in any sample. It is possible to characterize variations such as isoforms of miRNAs (isomirs) with single-base resolution, as well as analyse any small RNA or miRNA without prior sequence or secondary structure information. So, this technique allows to capture the complete range of small RNA and to identify new possible biomarkers.
Typical RNA-Seq experiment consists of isolating RNA, converting it to complementary DNA (cDNA), preparing the sequencing library, and sequencing it on an NGS platform.
Because small RNAs are lowly abundant, short in length (15–30 nt), and
lack polyadenylation, a separate strategy is often preferred to profile these
RNA species. To extract the miRNA fragments from the total RNA, a size
selection is performed: the total RNA is run on an agarose gel and the band
corresponding to the size of miRNAs is cut out for further processing. This
procedure excludes all bigger fragments, including all mRNAs and also rRNAs
from the samples. In the next step, the sequencing adapters are
ligated to the size-selected RNA molecules, followed by reverse transcription
to cDNA. The thus obtained cDNA library is run on an agarose gel again.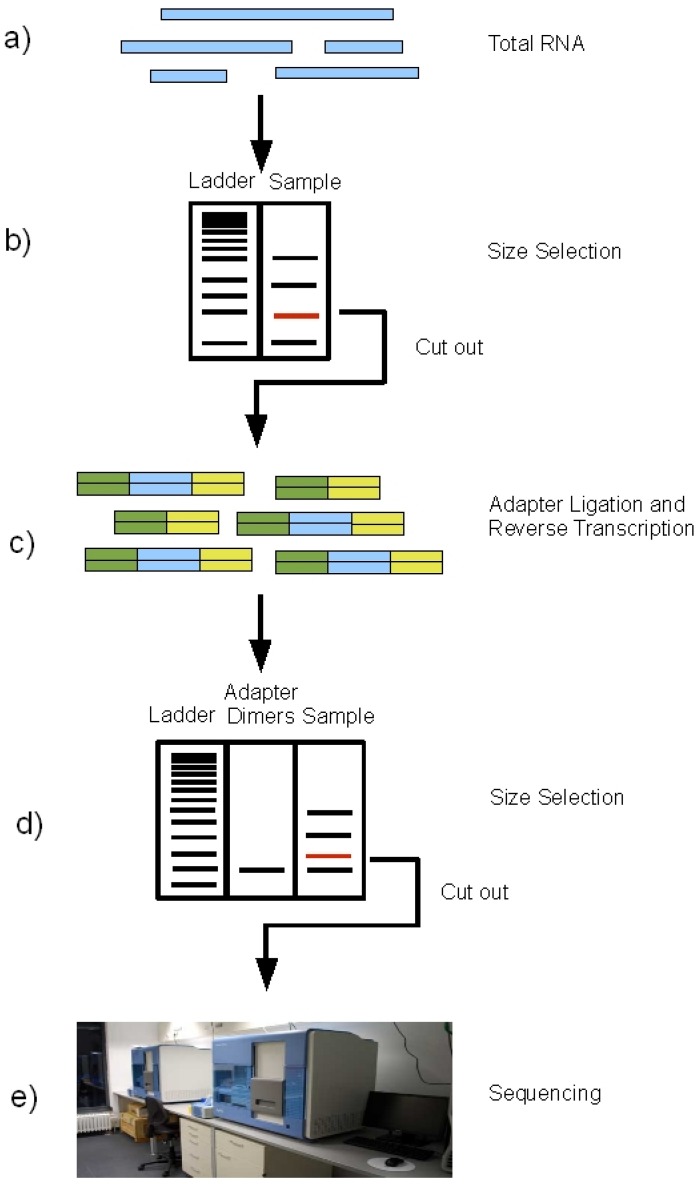
From Fig. 2, S.Motameny, 2010 (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3960865/figure/figure2/)
The output of a next generation miRNA sequencing experiment will typically contain millions of short reads. All produced reads are aligned to the reference genome of the sequenced organism and only perfectly matches are kept as potential miRNA reads. The remaining reads are discarded from further analysis. After the abundances of miRNAs are quantified for each sample, their expression levels can be compared between samples. In this way it is possible to identify miRNA that are preferentially expressed that particular time points, or in particular tissues or disease states. After normalizing for the number of mapped reads between samples, one can use a host of statistical tests to determine differential expression.
Another approach is to identify a miRNA’s mRNA targets, to understand genes whose expression they regulate. Public databases provide predictions of miRNA targets. Besides, since microRNAs are important regulators of many cellular processes such as survival, proliferation, and differentiation, they are usually involved in various aspects of cancer through the regulation of oncogene and tumor suppressor gene expression. In combination with the development of high-throughput profiling methods, miRNAs have been identified as biomarkers for cancer classification, response to therapy, and prognosis.
(Cecilia Thairi)
Nanopore sequencing
Nanopore systems offer real-time, scalable, direct DNA sequencing, in which the user chooses fragment length and the nanopore sequences the entire fragments. Using nanopore sequencing, a single molecule of DNA or RNA can be sequenced without the need for PCR amplification or chemical labeling of the sample. This technology is based on use of electrophoresis to transport an unknown sample through an orifice of 10−9 meters in diameter that is included in a Nanopore. A nanopore is located in a specific membrane and system always contains an electrolytic solutions, when a constant electric field is applied, an electric current can be observed in the system. The magnitude of the electric current density across a nanopore surface depends on the nanopore's dimensions and the composition of DNA or RNA that is occupying the nanopore. Samples cause characteristic changes in electric current density across nanopore surfaces, so it is possible recognize the specific nucleotides that compose the sample examining these alterations: each nucleotide has its specific profile of electric current alteration.
Substancially nanopore is set in an electrically resistant membrane. If an analyte passes through the pore or near its aperture, this event creates a characteristic disruption in current. Measurement of that current makes it possible to identify the molecule in question.
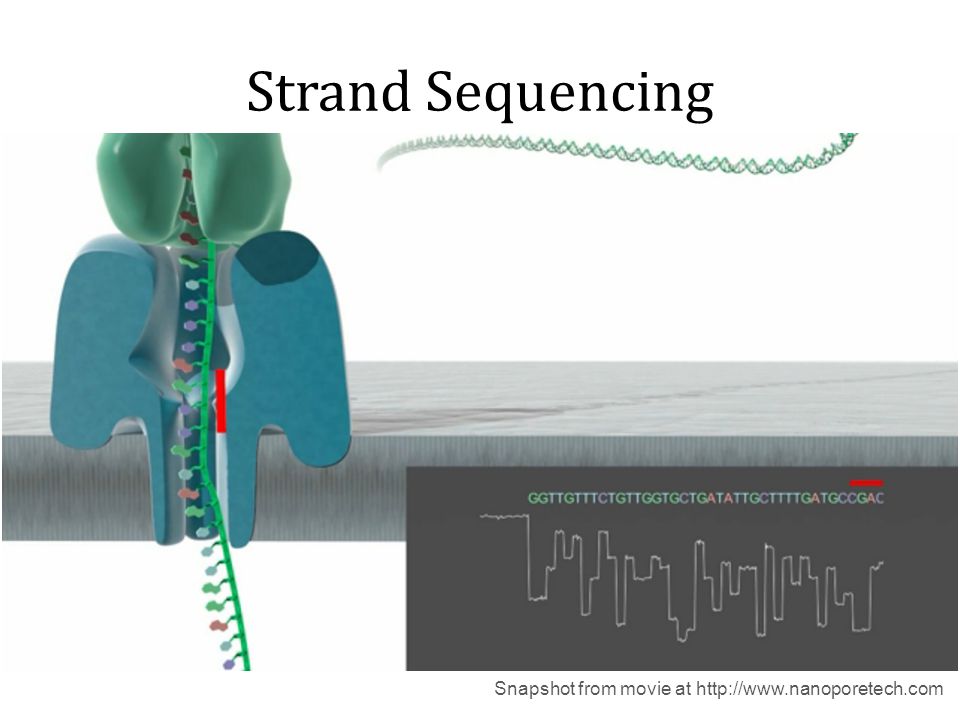
Holes can be created by proteins puncturing membranes (biological nanopores) or in solid materials (solid-state nanopores).
Biological nanopore sequencing relies on the use of transmembrane proteins, called porins, that are embedded in lipid membranes, alpha hemolysin (αHL), a nanopore from bacteria that causes lysis of red blood cells, is advantageous to identify specific bases moving through the pore: effectiveness can be improved with coupling of an exonuclease onto the αHL pore. The enzyme would periodically cleave single bases, enabling the pore to identify successive bases. Mycobacterium smegmatis porin A (MspA) is the second biological nanopore currently being investigated for DNA sequencing. The MspA pore has been identified as a potential improvement over αHL due to a more favorable structure because the natural nanopore was modified to improve translocation by replacing three negatively charged aspartic acids with neutral asparagines.
Solid state nanopore sequencing approaches do not incorporate proteins into their systems. Instead, solid state nanopore technology uses various metal or metal alloy substrates with nanometer sized pores that allow DNA or RNA to pass through. Measurement of electron tunneling through bases as ssDNA translocates through the nanopore is an improved solid state nanopore sequencing method, most research has focused on proving bases could be determined using electron tunneling. These studies were conducted using a scanning probe microscope as the sensing electrode, and have proved that bases can be identified by specific tunneling currents.
(Daniele Garelli)
Pacific Bio long-run sequencing
Single-molecule real-time sequencing developed by Pacific BioSciences offers longer read lengths and faster runs than the second-generation sequencing (SGS) technologies, making it well-suited for unsolved problems in genome, transcriptome, and epigenetics research. The highly-contiguous de novo assemblies using PacBio sequencing can close gaps in current reference assemblies and characterize structural variation (SV) in personal genomes. With longer reads, we can sequence through extended repetitive regions and detect mutations, many of which are associated with diseases. Moreover, PacBio transcriptome sequencing is advantageous for the identification of gene isoforms and facilitates reliable discoveries of novel genes and novel isoforms of annotated genes, due to its ability to sequence full-length transcripts or fragments with significant lengths. Additionally, PacBio’s sequencing technique provides information that is useful for the direct detection of base modifications, such as methylation. In addition to using PacBio sequencing alone, many hybrid sequencing strategies have been developed to make use of more accurate short reads in conjunction with PacBio long reads. In general, hybrid sequencing strategies are more affordable and scalable especially for small-size laboratories than using PacBio Sequencing alone. The advent of PacBio sequencing has made available much information that could not be obtained via SGS alone.
Mechanism and performance
PacBio sequencing captures sequence information during the replication process of the target DNA molecule. The template, called a SMRTbell, is a closed, single-stranded circular DNA that is created by ligating hairpin adaptors to both ends of a target double-stranded DNA (dsDNA) molecule. When a sample of SMRTbell is loaded to a chip called a SMRT cell, a SMRTbell diffuses into a sequencing unit called a zero-mode waveguide (ZMW), which provides the smallest available volume for light detection. In each ZMW, a single polymerase is immobilized at the bottom, which can bind to either hairpin adaptor of the SMRTbell and start the replication. Four fluorescent-labeled nucleotides, which generate distinct emission spectrums, are added to the SMRT cell. As a base is held by the polymerase, a light pulse is produced that identifies the base. The replication processes in all ZMWs of a SMRT cell are recorded by a “movie” of light pulses, and the pulses corresponding to each ZMW can be interpreted to be a sequence of bases (called a continuous long read, CLR). The latest platform, PacBio RS II, typically produces sequencing movies 0.5–4 h in length. Because the SMRTbell forms a closed circle, after the polymerase replicates one strand of the target dsDNA, it can continue incorporating bases of the adapter and then the other strand. If the lifetime of the polymerase is long enough, both strands can be sequenced multiple times (called “passes”) in a single CLR. In this scenario, the CLR can be split to multiple reads (called subreads) by recognizing and cutting out the adaptor sequences. The consensus sequence of multiple subreads in a single ZMW yields a circular consensus sequence (CCS) read with higher accuracy. If a target DNA is too long to be sequenced multiple times in a CLR, a CCS read cannot be generated, and only a single subread is output instead. Because PacBio sequencing takes place in real time, kinetic variation interpreted from the light-pulse movie can be analyzed to detect base modifications, such as methylation.
Review: PacBio Sequencing and Its Applications, Anthony Rhoads, Kin Fai Au, October 2015.
(Chiara Ossola)
Targeted sequencing and exome sequencing
New and improved methods and protocols have been developed to support a diverse range of applications of NGS, including the analysis of genetic variation. These methods aim to achieve ‘targeted enrichment’ of genome subregions and are referred to “targeted sequencing”. By selective recover and subsequent sequencing of genomic loci of interest, costs and efforts can be reduced significantly compared with whole-genome sequencing.
Targeted enrichment can be useful in a number of situations where particular portions of a whole genome need to be analyzed.
Efficient sequencing of the complete ‘exome’ represents a major current application. The exome has been defined traditionally as the protein-coding region covering between 1 and 2% of the genome, depending on species. It also may be extended to target functional nonprotein coding elements (e.g., microRNA, long intergenic noncoding RNA, etc.) as well as specific candidate loci.
There are two main categories of exome capture technology: solution-based and array-based.
- In solution-based, whole-exome sequencing (WES), DNA samples are fragmented and biotinylated oligonucleotide probes (baits) are used to selectively hybridize to target regions in the genome. Magnetic streptavidin beads are used to bind to the biotinylated probes, the nontargeted portion of the genome is washed away, and the polymerase chain reaction (PCR) is used to amplify the sample, enriching the sample for DNA from the target region. The sample is then sequenced before proceeding to bioinformatic analysis.

- Array-based methods are similar except that the probes are bound to a high-density microarray. The array-based method was the first to be used in exome capture, but it has largely been supplanted by solution-based methods, which require less input DNA and are consequently potentially more efficient.
Advantages of exome sequencing:
· Identifies variants across a wide range of applications, including population, translational and cancer studies, as well as for identification of rare genetic variants that are presumably responsible for complex genetic diseases, such as Mendelian disorders.
· Achieves comprehensive coverage of coding regions;
· Provides a cost-effective alternative to whole-genome sequencing (4–5 Gb of sequencing per exome compared to ~90 Gb per whole human genome).
· Enables researchers to focus their resources on the genes most likely to affect phenotype and offers an accessible combination of turnaround time and price;
· Produces a smaller, more manageable data set for faster, easier analysis compared to whole-genome approaches. DNA libraries can be prepared in as little as 1 day and require only 4–5 Gb of sequencing per exome.
· Detects variants in coding exons, with the capability to expand targeted content to include untranslated regions (UTRs) and microRNA for a more comprehensive view of gene regulation.
Sources:
Warr, A., Robert, C., Hume, D., Archibald, A., Deeb, N., & Watson, M. (2015). Exome Sequencing: Current and Future Perspectives. G3: Genes|Genomes|Genetics, 5(8), 1543–1550. http://doi.org/10.1534/g3.115.018564 lang="en-gb" xml:lang="en-gb">
https://www.gatc-biotech.com/en/expertise/targeted-sequencing/exome-enrichment.html
lang="en-gb" xml:lang="en-gb">Mertes F, ElSharawy A, Sauer S, et al. Targeted
enrichment of genomic DNA regions for next-generation sequencing. Briefings in Functional Genomics. 2011;10(6):374-386. doi:10.1093/bfgp/elr033.
(Marta Forcella)