Long-range chromatin interactions: 3C and derivatives up to Hi-C
(Restore this version)
Modified: 23 March 2018, 8:33 PM User: Silvia Bianchi →
3C
Chromosome conformation capture (3C) technology and its derivatives have been widely used to analyze the spatial organization of chromatin in a cell. These methods quantify the number of interactions between genomic loci that are nearby in 3-D space, but may be separated by many nucleotides in the linear genome. It can be applied to investigate the nuclear juxtaposition of any two genomic regions, in cis or trans. Such interactions may result from biological functions, such as promoter-enhancer interactions, or from random polymer looping, where undirected physical motion of chromatin causes loci to collide. 3C provides information on 3D chromatin structures that occur in living cells.
The protocol involves formaldehyde cross-linking of chromatin fragments and proteins in proximity followed by chromatin isolation and digestion with a restriction enzyme. The specific enzymes used are chosen in order to free a known or predicted DNA-DNA interaction mediated by a protein complex. The freed fragments are then ligated into rings and the crosslinks are reversed and the DNA is purified. Proteins are removed through the exposition to high ionic strands or high temperature. Both end-point PCR and quantitative real-time PCR (qPCR) can be employed to quantify the abundance of purified DNA 3C ligation products. The abundance of these recombinant fragments directly correlates to the interaction frequency of the two ligated regions.
This basic principle can be combined with other technologies to increase scale or specificity of the DNA loops being interrogated.
The derivative techniques of 3C are:
- Circularized Chromosome Conformation Capture (4C)
- Carbon Copy Chromosome Confromation Capture (5C)
- Chromatin interaction analysis by paired-end tag sequencing (CHIA-PET)
- ChIP-loop
- Hi-C
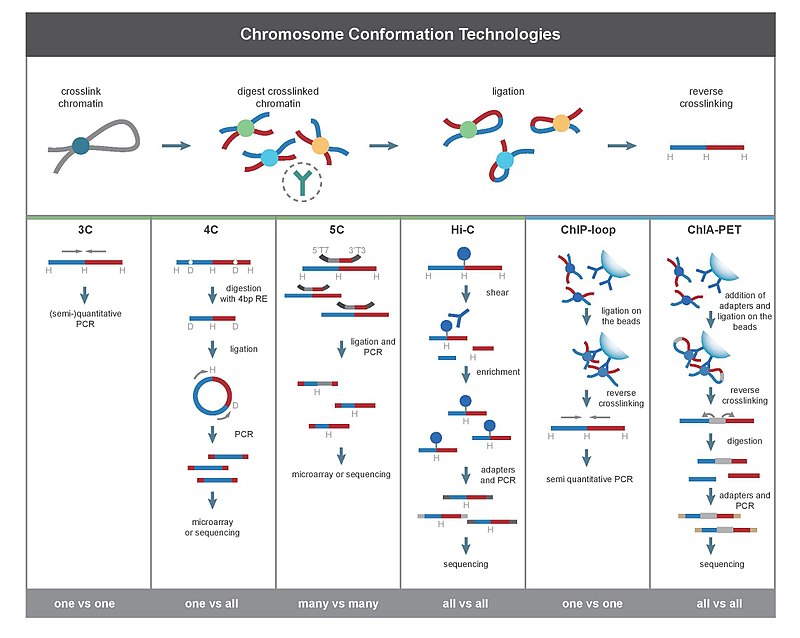
(Elisa Damo)
4C (Samuele Irudal)
As told before, 4C means Circularized Chromosome Conformation Capture. The first part of the protocol is equal to the one for basic 3C, so it will be reported without modification from the previous entry (thanks to the author).
The protocol involves formaldehyde cross-linking of chromatin fragments and proteins in proximity followed by chromatin isolation and digestion with a restriction enzyme. The specific enzymes used are chosen in order to free a known or predicted DNA-DNA interaction mediated by a protein complex.The freed fragments are then ligated into rings and the crosslinks are reversedand the DNA is purified. Proteins are removed through the exposition to high ionic strands or high temperature.
Following, these ligated segments are treated with another RE (a 4 pb RE), to create sticky ends: so, the two extremities will be able to interact with each other providing circularization. PCR is then carried out with divergent primers, preferentially recognising the contact points of the two fragments; sequencing can be managed after this passage.
ChIP-3C (or Chip-loop)
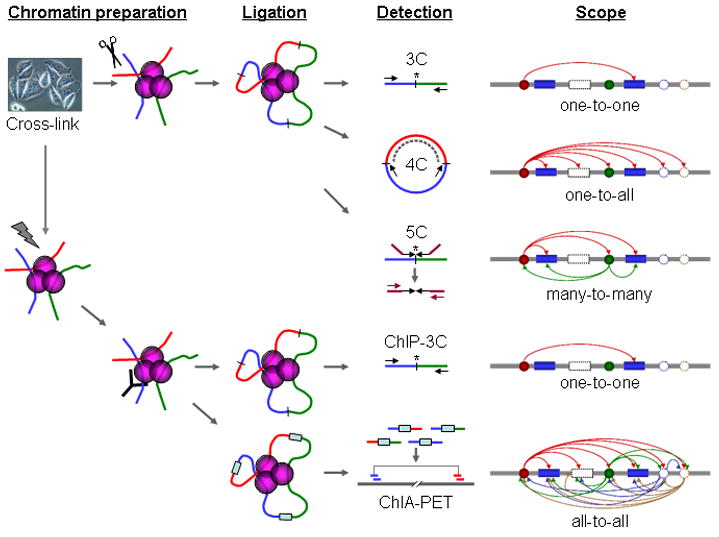
Fig 1. Schematic comparison of 3C, 4C, 5C, ChIP-3C, and ChIA-PET
ChIP-3C has been developed for a more specific identification of chromatin interactions, it detects DNA-DNA interactions mediated by a specific protein of interest. ChIP-3C is the result of combination between ChIP to the 3C protocol with the goal to reduce the non-specific noise that characterizes both these methods. The main advantage of ChIP-Loop is it reduces the background noise in 3C experiments and increases the specificity by selecting for a known protein mediating the DNA-DNA interaction. A reduction in background noise over a simple 3C assay is achieved by removing a large amount of genomic DNA by IP. Further, by targeting analysis to a specific protein of interest only specific, biologically relevant interactions are detected.
ChIP-3C has yielded insights into chromatin looping as mechanisms whereby important proteins can mediate cellular functions such as gene regulation. ChIP and 3C were first combined in 2005 to study a mouse model of Rhett. These researchers found that formation of a silent-chromatin loop is important for the function of the Dlx50Dlx6 locus. Further, they found that this loop was mediated by MECP2 and that this interaction is lost in individuals with Rhett Syndrome.
There are a few different ChIP-3C protocols in use, one of these involves:
1) preparation of urea ultracentrifugation-purified
2) restriction enzyme-digested
3) cross-linked chromatin
4) ChIP enrichment
5) proximity ligation
6) reverse crosslinking to free DNA fragments from protein binding
7) detection using PCR
While another omitted the urea ultracentrifugation purification and simply combined 3C and ChIP:
1) cells are are formaldehyde cross-linked followed by chromatin isolation and restriction digestion
2) Cross-linked chromatin is immunoprecipitated using an antibody against a protein of interest
3) he sticky DNA fragment ends are ligated while the chromatin fragments are still bound to the antibody,
4) The cross-links are reversed and the DNA is purified. A specific DNA-DNA interaction of interest can then be detected using PCR
However, the same final result is that predicted DNA-DNA interactions that are
mediated by a protein of interest are confirmed.
Another variant on ChIP-3C is the so-called 6C technique, which uses a cloning approach for detection, instead of site-specific PCR employed in conventional ChIP-3C.
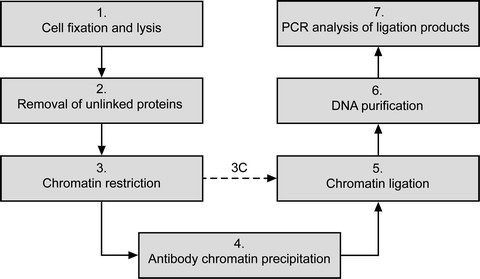
Fig 2. The main steps of ChIP-loop assay
One complication of is the accurate quantification of interaction levels, which must take into account both ChIP enrichment of the sites as well as high levels of non-specific chromatin noise due to random. Because 3C contains much noise, and does not include any steps to separate specific interactions from non-specific interactions before detection; therefore, if anyone uses ChIP to pull down specific protein-bound chromatin interactions from such 3C chromatin fragments, one would also co-precipitate the non-specific chromatin fragments that are attached to the specific interaction complexes. As such, it is likely that the use of the standard 3C protocol with the addition of ChIP just as it is would lead to high levels of false positives.
Minimize ligation bias by having sufficient starting material. By performing the ligation when the DNA is concentrated in a small volume, undesirable cross-ligation can occur between chromatin fragments. Validate with ChIP alone if possible. Cross-referencing ChIP-loop with ChIP data greatly reduces false-positives in the ChIP-loop assay. Use appropriate controls:
- Make a control templete that contains ligation products in equal amouts.
- Determine interaction frequency between known loci of increasing distance to estimate random interaction.
- When comparing two conditions, determine interaction frequency for a region expected to be the same in the two states.
Debate on the efficacy ChIP-3C rises questions on what is the nature of non-specific chromatin interactions, how such non-specific interactions are different from true and specific interactions, and how to experimentally separate non-specific chromatin interactions from true ones. The practical question is: can the non-specific and specific interactions be physically separated? If such a method can be found, the reduction in noise would greatly benefit the field of chromatin interactions.
(Riccardo Aucello)5C
What 5C is
5C (3C-Carbon Copy) is a novel 3C-based methodology for large-scale parallel detection of chromatin interactions.
5C was developed by Dostie and colligues in 2006 and the method was then validated, by the same group, by analyzing the human beta-globin locus and a conserved gene desert region located on human chromosome 16.
While, the 3C method tests the interaction between two known sites in the genome, 5C identifies all regions of interaction within a given genome domain. 3C is particularly suited for relatively small-scale studies, but large-scale mapping of chromatin interactions requires 3C libraries to be analyzed using a highthroughput detection method, such as microarrays or DNA sequencing. 5C answer to this request: it copy and amplify part of the 3C library, and then detection is made by microarrays or by quantitative DNAsequencing.
How 5C works
To copy in part the 3C library, 5C uses highly multiplexed ligation-mediated amplification (LMA): an amplification with primer pairs that anneal next to each other on the same DNA strand; it’s possible to reach high levels of multiplexing using thousands of primers in a single reaction.
To analyze chromatin interactions by 5C, a 3C library is first generated using the conventional 3C method. A mixture of 5C primers is then annealed onto the 3C library and ligated. Two types of 5C primers are used: 5C forward and 5C reverse primers. These primers are designed so that forward and reverse primers anneal across ligated junctions of head-to-head ligation products present in the 3C library. 5C primers that are annealed next to each other are then ligated with Taq ligase. This step generates a 5C library, which is amplified with universal PCR primers that anneal to the universal tails of the 5C primers. Forward and reverse 5C primers are only ligated when both are annealed to a specific 3C ligation product. Therefore, the 3C library determines which 5C ligation products are generated and how frequently. This is important because the abundance of each specific ligation product in the library is a measure of the frequency of interaction of the two corresponding loci. As a result, the 5C library is a quantitative “carbon copy” of a part of the 3C library. Forward and reverse 5C primers are designed to contain a unique sequence corresponding to the sense and antisense strand of the 3’-ends of restriction fragments and also contain universal tails for amplification (T7 at the 5’-ends of forward primers and T3c at the 3’-ends of reverse primers). To facilitate ligation, all reverse primers are phosphorylated at their 5’-ends.

To analyze interactions between many restriction fragments, multiple forward and reverse primers are mixed together in the same multiplex 5C reaction. This 5C primer design allows simultaneous amplification of all potential interactions between all restriction fragments recognized by a forward primer and all those recognized by a reverse primer.

Example
5C was used to study the interactions involving the LCR and three diagnostic fragments in the β-globin locus. With a proper primer design is possible to detect looping interactions between each of the three sections of the LCR and the surrounding chromatin in parallel in a single experiment.
Advantages and disadvantages
A major advantage of 5C is the fact that interactions between multiple elements of interest and other genomic elements can be analyzed in parallel in a single experiment.
5C is potentially limited by the number of 5C primers that can be used in a single reaction (limited coverage).
Conclusions
5C technology is a novel extension of 3C that allows simultaneous quantification of interaction profiles of many such “fixed” elements in parallel in a single reaction followed by analysis on a custom-designed microarray or by direct quantitative sequencing. To do this, reverse 5C primers are designed for each fixed fragment of interest, and forward 5C primers are designed for all other restriction fragments. When 5C is performed at a similar level of multiplexing, e.g., using 10,000 5C primers in a single experiment up to 25 million distinct chromatin interactions can be detected in parallel involving up to 40 Mb of DNA.
Applications
Large-scale application of 5C will:
- provide information about relationships between genes and regulatory elements
- allow to identify novel regulatory elements
- generate dense interaction maps providing a global overview of the conformation of a given genomic region.
Chromatin Interaction Analysis by Paired-End Tag Sequencing (ChIA-PET) is a technique that incorporates chromatin immunoprecipitation (ChIP)-based enrichment, chromatin proximity ligation, Paired-End Tags, and High-throughput sequencing to determine de novo long-range chromatin interactions genome-wide.
AIMS -> ChIA-PET can be used to identify unique, functional chromatin interactions between distal and proximal regulatory transcription-factor binding sites and the promoters of the genes they interact with and to unravel the mechanisms of genome control during processes such as cell differentiation, proliferation, and development. By creating ChIA-PET interactome maps for DNA-binding regulatory proteins and promoter regions, we can better identify unique targets for therapeutic intervention (Fullwood & Yijun, 2009).
The ChIA-PET method combines ChIP-based methods (Kuo & Allis, 1999), and Chromosome conformation capture (3C), to extend the capabilities of both approaches, that suffer from limitations when used independently to identify de-novo long-range interactions genome wide.
- ChIP-Seq, typically used for genome-wide identification of TFBS (Barski et al., 2007; Wei et al., 2006), provides only linear information of protein binding sites along the chromosomes (but not interactions between them), and can suffer from high genomic background noise (false positives).
- 3C, capable of analyzing long-range chromatin interactions, cannot be used genome wide and, like ChIP-Seq, also suffers from high levels of background noise. The noise increases in relation to the distance between interacting regions (max 100kb), therefore laborious and tedious controls are required for accurate characterization of chromatin interactions (Dekker et al., 2006).
The ChIA-PET method successfully resolves the issues of non-specific interaction noise found in ChIP-Seq by sonicating the chip fragments in order to separate random attachments from specific interaction complexes. The next step, which is referred to as enrichment, reduces complexity for genome-wide analysis and adds specificity to chromatin interactions bound by pre-determined TFs (transcription factors). The ability of 3C approaches to identify long-range interactions is based on the theory of proximity ligation. In regard to DNA inter-ligation, fragments that are tethered by common protein complexes have greater kinetic advantages under dilute conditions, than those freely diffusing in solution or anchored in different complexes. ChIA-PET takes advantage of this concept by incorporating linker sequences onto the free ends of the DNA fragments tethered to the protein complexes. In order to build connectivity of the fragments tethered by regulatory complexes, the linker sequences are ligated during nuclear proximity ligation. Therefore, the products of linker-connected ligation can be analyzed by ultra-high-throughput PET sequencing and mapped to the reference genome. Since ChIA-PET is not dependent on specific sites for detection as 3C and 4C are, it allows unbiased, genome-wide de-novo detection of chromatin interactions (Fullwood et al., 2009).
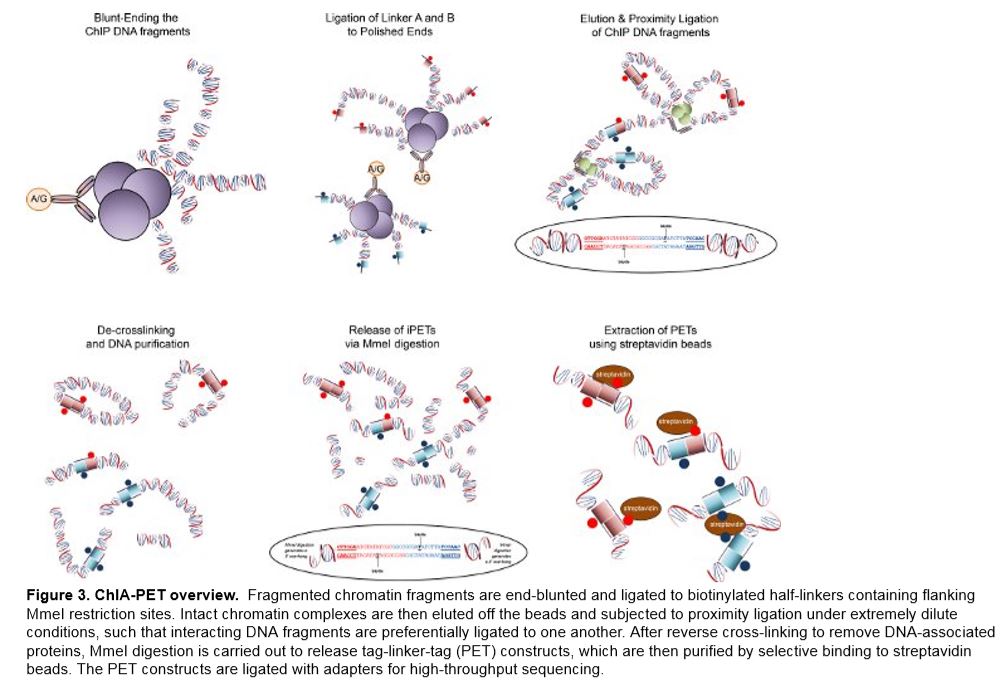
Strengths and weaknesses
Advantages of the ChIA-PET method:
-> ChIA-PET has a potential to be an unbiased, whole-genome and de-novo approach for long-range chromatin interaction analysis (Fullwood & Yijun, 2009).
-> A ChIA-PET experiment is capable of providing two global datasets: The protein factor binding sites (self-ligated PETs); and The interactions between the binding sites (inter-ligated PETs).
-> ChIA-PET involves ChIP to reduce the complexity for genome-wide analysis and adds specificity to chromatin interactions bound by specific factors of interest.
-> ChIA-PET is compatible with tag-based next-generation sequencing approaches such as Roche 454 pyrosequencing, Illumina GA, ABI SOLiD, and Helicos.
-> ChIA-PET is applicable to many different protein factors involved in transcriptional regulation or chromatin structural conformation.
-> ChIA-PET analysis can be applied to chromatin interactions involved in a particular nuclear process. By using general TFs such as RNA Polymerase II, it may be possible to identify all chromatin interactions involved in transcription regulation. Further, the use of protein factors involved in DNA replication or chromatin structure would allow identification of all interactions due to DNA replication and chromatin structural modification (Fullwood et al., 2009).
Weaknesses of the ChIA-PET method:
-> It is well established that cis and trans-regulatory complexes contain unique combinations of proteins based on cell and tissue specific conditions (Dekker et al., 2006). While identification of single, functional TFBS is a significant advancement, the use of ChIA-PET to identify individual proteins in a complex would require guess work and multiple experiments to identify each interacting protein. This would be a costly and time-consuming endeavour.
-> ChIA-PET is limited by the quality, purity, and specificity of the antibodies used (Fullwood et al., 2009).
-> ChIA-PET is dependent on identification of sequences that can be mapped to the reference sequence (ref).
-> ChIA-PET requires the use of peak-calling computer algorithms to organize and map PET reads to the reference genome. Because of variations between software platforms, results can vary depending on which program is used.
-> Although repetitive DNA regions can be associated with gene regulation (Polak & Domany, 2006), they need to be removed as they can affect the data (Fullwood et al., 2009)
Source:
Wikipedia
Goh Y. et al., Chromatin Interaction Analysis with Paired-End Tag Sequencing (ChIAPET) for Mapping Chromatin Interactions and Understanding Transcription Regulation.
(Laura Tasca)
Hi-C
Hi-C is an extension of 3C that is capable of identifying long range interactions in an unbiased, genome-wide fashion.
In Hi-C, cells are fixed with formaldehyde, causing interacting loci to be bound to one another by means of covalent DNA-protein cross-links. When the DNA is subsequently fragmented with a restriction enzyme, these loci remain linked. A biotinylated residue is incorporated as the 5' overhangs are filled in. Next, blunt-end ligation is performed under dilute conditions that favor ligation events between cross linked DNA fragments. This results in a genome-wide library of ligation products, corresponding to pairs of fragments that were originally in close proximity to each other in the nucleus. Each ligation product is marked with biotin at the site of the junction. The library is sheared, and the junctions are pulled-down with streptavidin beads. The purified junctions can subsequently be analyzed using a high-throughput sequencer, resulting in a catalog of interacting fragments.
Direct analysis of the resulting contact matrix reveals numerous features of genomic organization, such as the presence of chromosome territories and the preferential association of small gene-rich chromosomes.
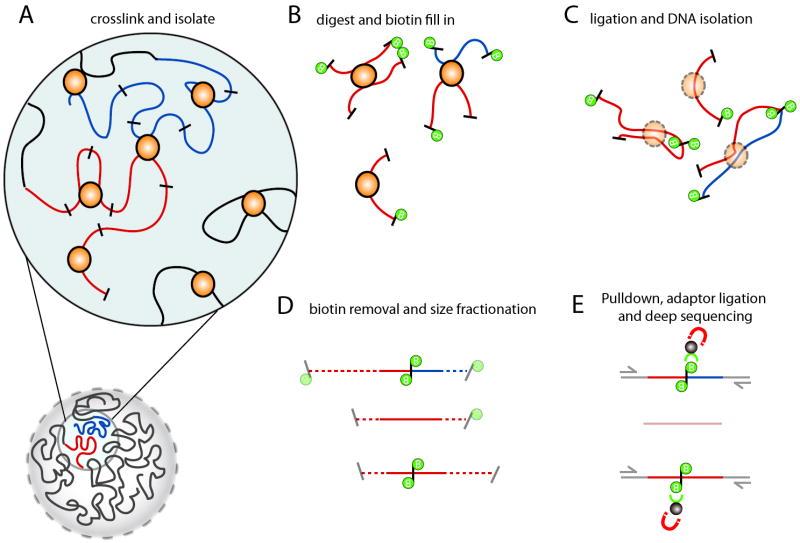
Figure 1. Overview of
Hi-C technology
A) Hi-C detects chromatin interaction both within and between chromosomes by covalently
crosslinking protein/DNA complexes with formaldehyde. B) The chromatin is digested with
a restriction enzyme and the ends are marked with a biotinylated nucleotide. C) The DNA in
the crosslinked complexes are ligated to form chimeric DNA molecules. D) Biotin is
removed from the ends of linear fragments and the molecules are fragmented to reduce their
overall size. E) Molecules with internal biotin incorporation are pulled down with
streptavidin coated magnetic beads and modified for deep sequencing. Quantitation of
chromatin interactions is achieved through massively parallel deep sequencing.
The Hi-C method produces genome-wide interaction maps. As such, Hi-C provides a unique and powerful tool to study nuclear organization and, chromosome architecture. Thus, Hi-C data will add a spatial context to biological inquires and will facilitate the discovery of the fundamentals of gene regulation, nuclear partitioning, and the biophysics of chromatin dynamics.
Hi-C does not capture the fine detail of subnuclear compartments like electron microscopy nor can it measure the dynamics of interactions between multiple genomic loci like fluorescent microscopy. It does, however provide the ultimate connectivity between the genomic sequence and spatial conformation.
The genome-wide power and versatility of Hi-C makes it ideal for the study of the basic biology of genome organization and its implications for health and disease.
(Silvia Bianchi)