FISH
(Restore this version)
Modified: 20 March 2018, 9:10 PM User: Fabiola Varese →
FISH (Giada Cipollina)
Fluorescent in situ hybridization (FISH) is a molecular cytogenetic technique used to detect the presence of a specific DNA sequence on a chromosome.
The technique is based on the use of a probe: a short DNA sequence (10-25 nucleotides), specifically designed for the target sequence. To allow the detection of a wanted genic locus the probe is fluorescent-labelled (direct labelling) or can be rendered fluorescent in a subsequent phase (indirect labelling).
The first protocol step is the attachment of either interphasic or metaphasic chromosomes on a solid surface (glass); then the sample and probe solution is denatured and incubation starts. During the incubation period (hours, depending on the protocol), target DNA and probe interact and hybridize. To avoid background, unbound/partially bound probes are washed away, by repeated washes, after incubation.
The detection step can be different: in case of direct detection the probe is already fluo-marked (A in scheme below) and the sample can be directly observed at fluorescence microscope. On the contrary, in the indirect labelling it is necessary to add an enzymatic or immunological detection system, which will render the probe fluorescent. This method needs more time but allows to amplify the signal and get a better detection. For example an indirect labeling is based on tagging the probe with biotin (grey symbol in figure B below); after DNA-probe hybridization streptavidin-fluorescent dye (light green and yellow symbol in figure below) is added and in this way is possible to detect the signal.

FISH is a versatile technique: can be useful to detect a gene locus on a chromosome, chromosomal abnormalities and rearrangements. In clinics FISH is therefore used to diagnose several disease, including some cancers.
Nowadays several connected techniques have been developed starting form the “classical” FISH, which is focused on chromosomal loci.
Figure references:
(modified by) http://www.wikiwand.com/en/Chromogenic_in_situ_hybridization
(Giada Cipollina)
Immuno-FISH (Fabiola Varese)
As the name suggests, this is a combination of two techniques, the standard FISH, either on flattened chromosome preparations (2-D FISH) or on three-dimensionally preserved nuclei (3-D FISH), and indirect or direct immunofluorescence. Immunofluorescence permits the detection of nuclear proteins (modified histones, histone variants and modifiers, transcription machinery and factors, nuclear sub-compartments, etc), so that both DNA and proteins can be analyzed on the same sample. Numerous methods involving a variety of fixation and permeabilization techniques can be used for immunofluorescence applications, and the choice depends on cell type, epitope, and antibody being used. The choice of fluorochrome to which the secondary antibody is conjugated will depend on the fluorochrome with which the FISH probe is labelled.
The combination of these techniques is often used to investigate co-localization of genomic regions with proteinaceous entities within interphase nuclei such as nucleoli or promyelocytic leukemia (PML) bodies. It has helped to position chromosomes in interphase nuclei.
One of the most used applications, that rquires an appropriate microscopy method (confocal microscopy would be the best) is the 3D-immuno-FISH, useful to map in a three-dimensional way DNA and proteins in the nucleus, preserving their spatial positions. The main difficulty is to preserve the spatial arrangement of chromatin while making DNA accessible for probes.
Some protocols establish that FISH procedure comes first, but not all antigens will be preserved after the various steps in the protocols. So, for unstable antigens that do not withstand FISH pretreatments (B and C in the figure), primary and secondary antibodies can be applied followed by a post-fixation step prior to the FISH procedure. For robust antigens, the addition of antibodies can follow hybrid-detection.
An important step, in both cases, is the counterstaining, used to test the preservation of nuclear morphology and to select well-preserved nuclei to use in further analysis. The counterstaining dye is chosen depending on the fluorochromes used and on the wavelengths available for microscopy. The most used is the DNA-specific DAPI (producing a blue fluorescence, but there are also dyes that stain both DNA and RNA, so, preliminary digestion with RNase before DNA staining is required for DNA-immuno-FISH.

(Fabiola Varese)
RNA-FISH (Luca Torello)
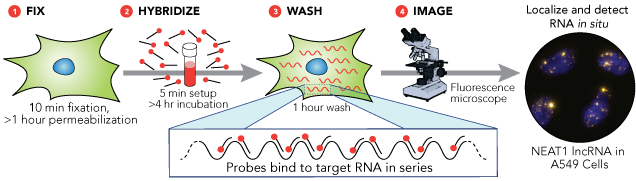
As we already said, FISH is a very powerful method to detect optically specific nucleic acid sequences (both DNA and RNA sequences) in fixed but otherwise intact cells. DNA FISH is useful to understand the number of gene copies, the location of the gene in the nucleus when it is turned on/off, DNA repair and replication studies. However, RNA-FISH is a technique that has become a very powerful tool to study gene expression and RNA biology. In fact, RNA-FISH is a quantifiable method that can determine the localization of single mRNA molecules in the cell, their number, their transcription and degradation rates and more.
There are two categories of RNA-FISH: those that use some form of signal amplification, and those that rely on direct detection of a signal.
Direct detection involves labeling the probes themselves with fluorophores (the probes must have enough fluorescence to be detectable above background autofluorescence). One technique is to use a set of short single-stranded DNA oligonucleotides complementary to various regions of the target RNA, each labelled with one or more fluorescent moieties. The binding of multiple probes enables the localisation of the target RNA via fluorescence microscopy as a fluorescent spot. The advantage of this approach is that the off-target binding of a single oligonucleotide in the probe pool will either be undetectable or readily distinguishable to the much brighter spots corresponding to the true RNA, thus reducing the chances of false positives. False negatives are similarly unlikely, for even if a single probe out of the pool fails to bind, the rest are likely to bind.

In order to circumvent the limitation of low signals from the relatively small numbers of fluorescent molecules targeted to mRNA in these direct detection methods (A), researchers have also developed a large variety of methods to amplify signals from individual molecules. Some of these are relatively simple extensions of the direct detection methods, such as detection of the probe by fluorescently labelled antibodies targeting specific haptens (small molecule that reacts with specific antibody but is not immunogenic by itself) incorporated in large numbers into an RNA probe (B). Others involve targeting nucleic acid probes with a single or few haptens with antibodies conjugated to enzymes; those enzymes in turn will act upon a substrate in such a manner as to create a fluorescent product that will become covalently linked to surrounding molecules (C).
Another technique is the RNAScope Assay (Figure Below). Here, we produce two unlabeled tandem probes, containing a short complementary region (18-25 bases), a spacer sequence and a 14-base tail sequence (rapresented as “Z” in the image). After hybridization with the target probes, comes a second hybridization step with a pre-amplifier probe. This is a long probe that contains a complementary sequence to the 28 bases of the two target probes tails (14+14). Therefore, only when the two target tails hybridize one next to the other the pre-amplifier will hybridize. The pre-amplifier contains 20 binding sites for an amplifier probe, which in turn contains 20 binding sites for the labelled probe. Thus, for each target probe pair, we get 20×20=400 labelled probes. Such method has the advantage of labeling targets to such a degree that the signals are easily visible even by eye, precluding the need for expensive optical setups. In addition, they are able to reliably detect short RNA molecules such as miRNA. However, such methods are somewhat prone to lower detection efficiencies owing to the large number of steps in such protocols, each of which has some probability of failure.

Figure References:
1. https://biosearchassets.blob.core.windows.net/assets/standard/PAGE/2239/stellaris-method.png
2. https://ars.els-cdn.com/content/image/1-s2.0-S1046202316300123-gr1.jpg
3. https://greenfluorescentblog.wordpress.com/2013/02/21/rnascope-a-novel-fish-in-the-sea/
(Luca
Torello)
Flow-FISH and Q-FISH (Elena Richiardone)
Fluorescence in situ hybridization (FISH) is a powerful cytogenetic technique that is used to detect and localize specific nucleic acid sequences in the cellular environment. In order to increase throughput, FISH can be combined with flow cytometry (flow-FISH) to enable the detection of targeted nucleic acid sequences technique and to quantify the copy number of specific repetitive elements in genomic DNA of whole cell populations via the combination of flow cytometry with cytogenetic fluorescent in situ hybridization staining protocols. FISH and flow-FISH methods are employed in a number of different applications and the utility of these methods has been successfully demonstrated in telomere length determination, cellular identification and gene expression, monitoring viral multiplication in infected cells, and bacterial community analysis and enumeration. Traditionally, the specificity of FISH and flow-FISH methods has been imparted by DNA oligonucleotide probes. Recently however, the replacement of DNA oligonucleotide probes with nucleic acid analogs as FISH and flow-FISH probes has increased both the sensitivity and specificity of each technique due to the higher melting temperatures (T(m)) of these analogs for natural nucleic acids. Locked nucleic acid (LNA) probes are a type of nucleic acid analog that contain LNA nucleotides spiked throughout a DNA or RNA sequence. When coupled with flow-FISH, LNA probes have previously been shown to outperform conventional DNA probes and have been successfully used to detect eukaryotic mRNA and viral RNA in mammalian cells.
Flow-FISH is a modification of another technique for analyzing telomere length, Q-FISH, that employs peptide nucleic acid probes of a 3'-CCCTAACCCTAACCCTAA-5' sequence labeled with a fluorescin fluorophore to stain telomeric repeats on prepared metaphase spreads of cells. Images of the resultant fluorescent spots could then be analyzed via a specialized computer program (method and software available from the Flintbox Network) to yield quantitative fluorescence values that can then be used to estimate actual telomere length. The fluorescence yielded by probe staining is considered to be quantitative because PNA binds preferentially to DNA at low ionic salt concentrations and in the presence of formamide, thus the DNA duplex may not reform once it has been melted and annealed to PNA probe, allowing the probe to saturate its target repeat sequence (as it is not displaced from the target DNA by competing anti sense DNA on the complementary strand), thus yielding a reliable and quantifiable readout of the frequency of PNA probe target at a given chromosomal site after washing away of unbound probe.

Unlike Q-FISH, Flow-FISH utilizes the quantitative properties of telomere specific PNA probe retention to quantify median fluorescence in a population of cells, via the use of a flow cytometer, instead of a fluorescence microscope. The primary advantage of this technique is that it eliminates the time required in Q-FISH to prepare metaphase spreads of cells of interest, and that flow cytometric analysis is also considerably faster than the methods required to acquire and analyze Q-FISH prepared slides. Flow-FISH thus allows for a higher throughput analysis of telomere length in blood leukocytes. The most recent versions of the flow-FISH technique include an internal control population of cow thymocytes with a known telomere length detected by TRF (telomere restriction fragment) analysis to which the fluorescence of a given unknown sample may be compared. Because cow thymocytes take up LDS751 dye to a lesser extent than their human counterparts, they may be reliably differentiated via plotting and gating the desired populations.

Main advantages are:
· Highly reproducible results
· Possible to analyze subsets of cells using physical and immunofluorescent markers.
· Better throughput than Q-FISH
· Possible to acquire fluorescence data on thousands of cells
(Elena Richiardone)
others....