RNA binding protein mapping: RIP, CLIP, HITS-CLIP, PAR-CLIP
(Restore this version)
Modified: 15 May 2019, 5:40 PM User: Anna Migni →
Return to index
RNA-binding proteins have crucial roles in different cellular processes, especially the ones concerning the post-transcriptional control of RNAs, such as splicing, polyadenylation and mRNA localization and translation. Detecting RBPs, which exhibit highly specific recognition of their RNA targets by recognizing their sequences and structures, is important in order to understand the mechanisms involved in development, cellular differentiation, metabolism, health and disease.
RIP
CLIP
[Anna Migni]
CLIP (cross-linking immunoprecipitation) is an antibody-based method used in order to analyse protein interactions with RNA or to precisely identify RNA modification sites.
CLIP differs from RIP in the use of UV radiation to cross-link RNA binding proteins to the RNA that they are bound to.
It is useful to understand the role of RNA in transcription, slicing, translation but also to study RNA interference and gene regulation by non-coding RNAs.
Basic Principle of CLIP: the use of UV light promote the formation of a covalent bond between proximal proteins and RNA. It occurs only at the sites of direct contact and it preserves RNA-protein interaction
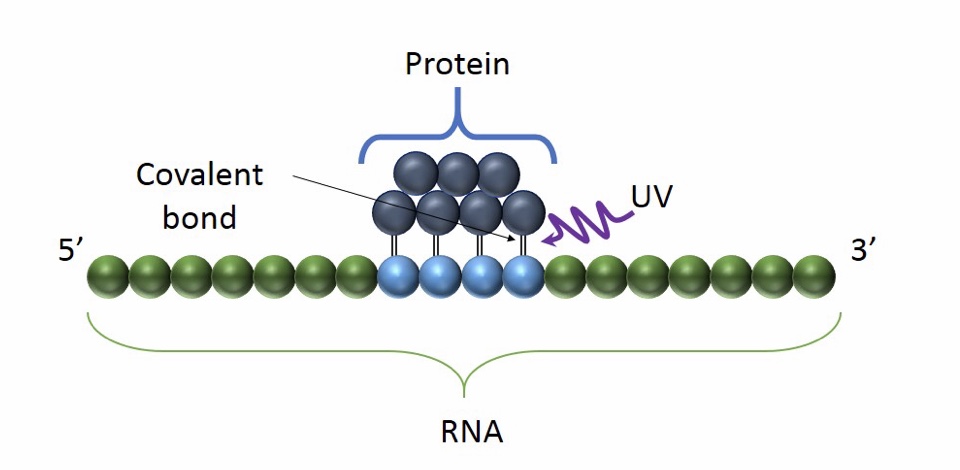
Advantages:
- The main advantage of CLIP is the high accuracy and specificity. UV cross-linking in fact, avoids protein-protein cross linking.
- Easy in the preparation of the samples
Limits:
- CLIP protocol requires a large numeber of cells and tissues.
- UV light may cause mutation in DNA.
- It requires a meticulous protocol, including bioinformatic analysis.
There are many variants of CLIP that can overcome the disadvantages and improve its features, depending on the aim.
Some esamples are PAR-CLIP, iCLIP, HITS-CLIP.
Workflow of iCLIP
Step 1: UV exposure to form covalent bound between proteins and RNA in direct contact. The cell is lisated and the protein of interest is detected by IP.
Step 2: washing to remove free RNA and RNA raptors are added at the 3’ ends.
Step 3: Proteinase K digestion
Step 4: RT PCR
Step 5: Adapters are ligated at 5’ cDNA ends via circularization. Then restriction enzyme cleavage is performed to linearised cDNA.
Step 6: sequencing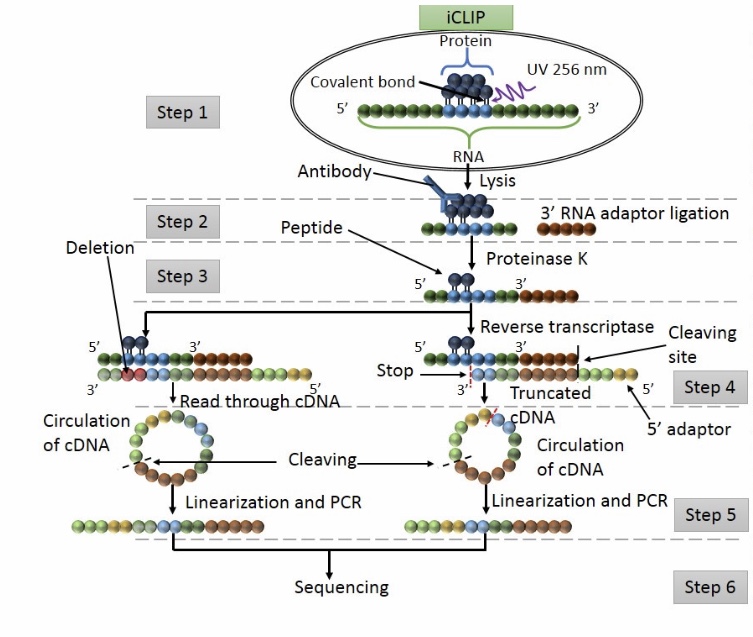
If you want to have more details about this protocol:
HITS-CLIP
[Vittoria Berutto, Arianna Rizzo]
High-throughput sequencing of RNA isolated by crosslinking immunoprecipitation (HITS-CLIP, also known as CLIP-Seq) was developed as a genome wide mean to map protein-RNA binding sites. HITS-CLIP capitalizes on in vivo UV crosslinking of cells or tissues to form covalent bond between RNAs and proteins that are in direct contact. This step is followed by an immunopurification of the RNA-protein complexes of interest and finally next-generation sequencing of the isolated RNA tags.
In order to overcome the limitations of CLIP experiments (the small number of tags that are obtained can’t give any robust generalization about the nature of the RNA-protein interactions), it has been applied next-generation high throughput sequencing methods to CLIP: so, we can talk about HITS-CLIP. Analysis of the same RNA-protein interactions with HITS-CLIP yielded over 1000-fold more unique tags. Given the large (and ever-increasing) amounts of data available using next-generation sequencing technologies, bioinformatic analysis of raw tags is an important aspect of such studies.
This new technique, which utilizes in vivo UV crosslinking technologies and next-generation sequencing, has caught the attention of the RNA community as a means of achieving a new depth of understanding about how protein-RNA complexes interactions regulate gene expression in living cells.
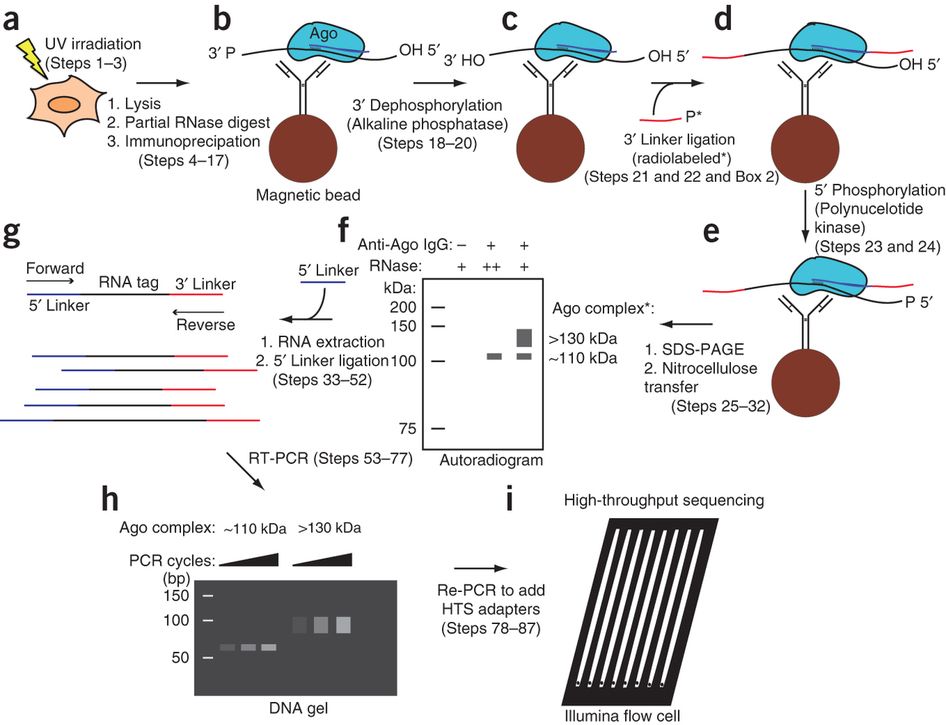
Workflow of HITS-CLIP:
(a) UV irradiation of live cells or tissue induces RNA-protein cross-links (Steps 1–3)(b) Material is lysed, RNA is partially digested, and the target protein is immunopurified with antibody coupled to magnetic beads (Steps 4–17)
(c) Alkaline phosphatase treatment removes 3′ hydroxyl to permit 3′ linker ligation (Steps 18–20)
(d) Radiolabeled (*) 3′ linker (red) is ligated to RNA tags (Steps 21 and 22)
(e) Polynucleotide kinase treatment phosphorylates 5′ RNA ends, allowing subsequent
5′ linker ligation (Steps 23 and 24)
(f) Complexes are eluted from beads and separated by SDS-PAGE. After transfer to nitrocellulose membrane, complexes are visualized by autoradiography (Steps 25–32). (g) RNA is extracted from the desired membrane region by protease treatment, and the 5′ linker (purple) is ligated to tags (Steps 33–52)
(h)
Tags are amplified by RT-PCR (Steps 53–77)
(i) After the addition of sequencing
adapters in a second PCR step, the samples are sequenced on the Illumina platform (Steps 78–87)
Advantages:
- Crosslinking stabilizes the protein-target binding
- UV crosslinking can be carried out in vivo
- Provides low background and higher resolution of binding site, due to RNase digestion
- No prior knowledge of the RNA is required
- Genome-wide RNA screen
Disadvantages:
- Over-representation of the RT complement due to mispriming
- Antibodies not specific to the target may precipitate nonspecific complexesU
- UV crosslinking is not efficient and requires close protein-RNA interactions
- Artifacts may be introduced during the crosslinking process
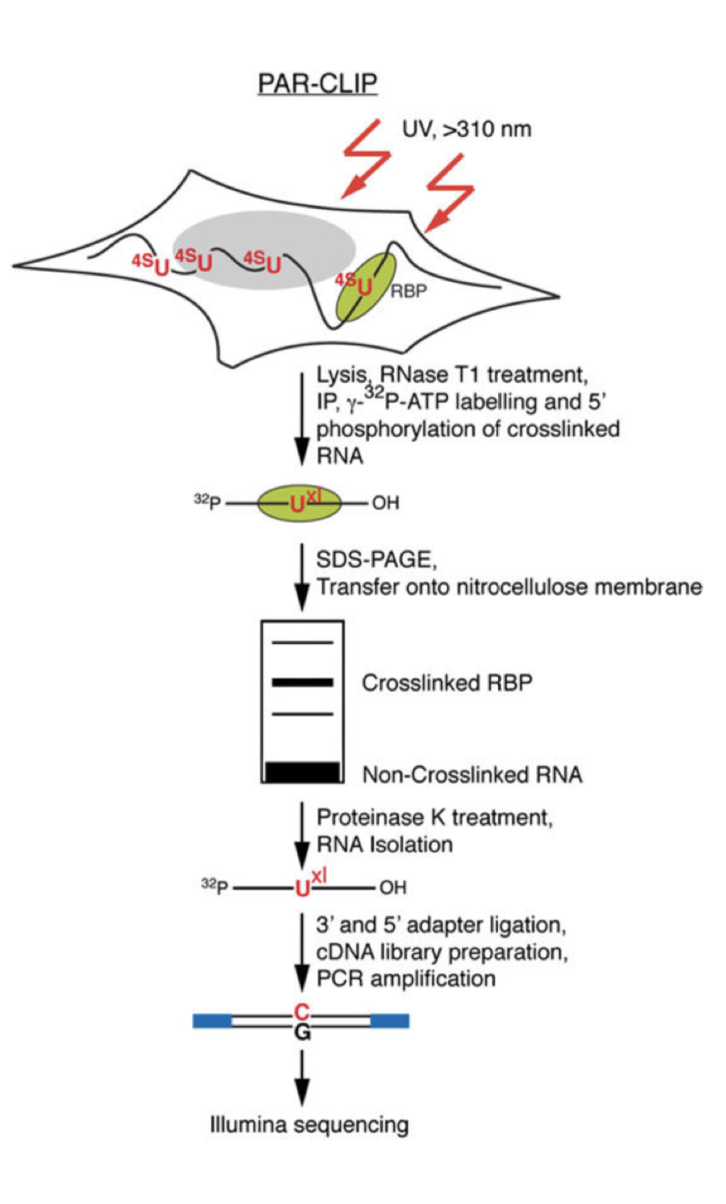
PAR-CLIP
[Audisio Alice]
Photoactivatable-Ribonucleoside-Enhanced Cross linking and Immunoprecipitation
PAR-CLIP is a method for identifying RBP binding sites on target RNAs with nucleotide-level resolution.
It's applicable to any protein that contact directly RNA, including RBPs that are predicted to bind in a sequence- or structure- dependent manner at discrete RNA recognition elements (RREs), and those that are thought to bind transiently, such as RNA polymerases or helicases.
Important features :
Photoactivatable ribonucleosides, usually the 4-thiouridine (4SU) and more rarely the 6-thioguanine (6SG), are incorporated into nascent RNA transcripts.
- The labeled RNAs are excited in living cells with UVA or UVB light (>310 nm) and yield photoadducts with interacting RBPs, that a represents an increased cross-linking efficiency compared to 254 nm CLIP
- A characteristic mutation (T-to-C for 4SU and G-to-A for 6SG) is introduced during reverse transcription at the position of cross-linking. This mutation permits the localization the sites of RNA–RBP interaction with nucleotide resolution and enables the user to computationally remove the ubiquitous background of co-purifying fragments of cellular RNAs that otherwise may be misinterpreted as signal
Method :
- Expanding Cells
- UV-cross-linking
- Cell Lysis and RNase T1 Digest
Immunoprecipitation and Recovery of Crosslinked Target RNA Fragments
- Preparation of Magnetic Beads
- Immunoprecipitation (IP), Second RNase T1 Digestion, and Dephosphorylation
- Radiolabeling of RNA Segments Crosslinked to Immunoprecipitated Proteins
- SDS Polyacrylamide Gel Electrophoresis, Transfer, and Recovery of RNA from Nitrocellulose Membrane
- Proteinase K Digestion
cDNA Library Preparation and Deep Sequencing
- 3′ Adapter Ligation
- 5′ Adapter Ligation
- Reverse Transcription
- PCR Amplification
- Optional: Determination of Incorporation Levels of 4-Thiouridine into Total RNA
PAR-CLIP Analysis
- Illumina sequencing : >200 million sequence reads per sample
- Sophisticated approaches to identify binding sites
- Calculation of the common sequence motifs of the RRE using one of the programs developed for the analysis of transcription-factor binding sites on DNA ( MEME, MDScan, PhyloGibbs, cERMIT, Gimsan)
Generally, the analysis of the sequence reads begins by alignment to the genome, allowing for at least one error (substitution, insertion, or deletion) to capture cross-linked reads with cross-linking-induced mutations.
Next, overlapping sequence reads are grouped, taking into account the frequency of cross-linking-induced mutations.
The frequency of the T-to-C mutations (or G-to-A mutations when using 6SG) allows ranking of groups to predict those interactions with the highest functional impact.