Transcription Factor mapping and prediction
(Restore this version)
Modified: 1 May 2019, 7:54 PM User: Valeria Bastianini →
Return to index
List of subjects:
1. TF binding site mapping genome-wide
2. TF sequence element prediction
3. biochemical determination of binding sequence (e.g. SELEX)
4. Activity assays for TFs
(Samar El Sherbiny)
1. TF binding site mapping genome-wide
The identification of transcription factor binding sites (TFBS) is an important initial step in determining the DNA signals that regulate transcription of the genome. We have different techniques able to identify this TFBS:
a. ChIP-seq
ChIP-seq is a technique that combines two aspects: chromatin immunoprecipitation with sequencing. It's a powerful method for identifying genome-wide DNA binding sites for transcription factors and other proteins. Following ChIP protocols, DNA-bound protein is immunoprecipitated using a specific antibody. The bound DNA is then coprecipitated, purified, and sequenced.
Advantages:
- We can analyze the entire genome;
- It reveals gene regulatory networks in combination with RNA sequencing and methylation analysis;
- It offers compatibility with various input DNA samples.
ChIP-seq workflow:
The first part is the Chromatin immunoprecipitation or library prep:
- Chromatin is crosslinked in order to fix all the interactions with proteins. Than it is fragmentated, for this step we can use different methods, but the most used is sonication.

- Than we have the enrichment, specific antibodies for the protein of interest are added to the sample in order to perform the immunoprecipitation.
- At this point we can revert the crosslinking in order to obtain the DNA that will be purified. In order to perform NGS for the following step, that is sequencing, we will need also additional steps like end repair, phosphorilation, A-tailing, logation to the adapter and finally our sample will be ready for the sequencing.
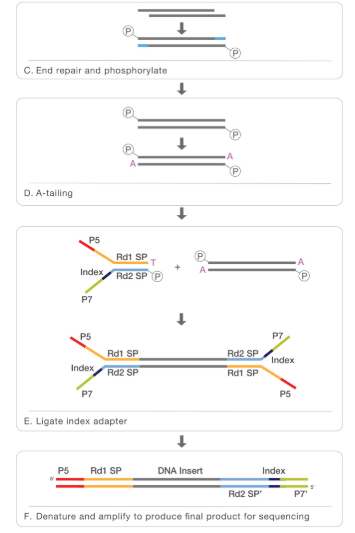
The second part is the sequencing:
Before sequencing the fragments are amplyfied thanks to PCR and than we can have the sequencing, usually performed with NGS. In order to understand better all the steps of NGS sequencing i put a video from Illumina web site: https://www.youtube.com/watch?time_continue=290&v=fCd6B5HRaZ8
After sequencing the third part of ChIP-seq technique is the analysis of data:
Basically we obtain reads, they are small fragments that we can obtain after the sequencing. All the reads are mapped on the reference genome and we can see an enrichment in some spots: they are called peaks and from this output we can assume that the most enriched sites are with high probability the sites in which we have the binding between the transcription factor and the DNA. An important step in order to obtain data that are precise, is to estimate and eliminate the background. If we want to know the motif we need to perform the peak calling and peak annotation. Usually the binding site is around the center of the peak. To calculate the motif we need bioinformatic tools and specific algorithms.
b. ChIP on chip
ChIP-on-chip is a technology that combines chromatin immunoprecipitation with DNA microarray. It used to investigate interactions between proteins and DNA in vivo. Specifically, it allows the identification of the binding sites, for DNA-binding proteins on a genome-wide basis. This assay can be divided into two main parts:
- Wet-lab step
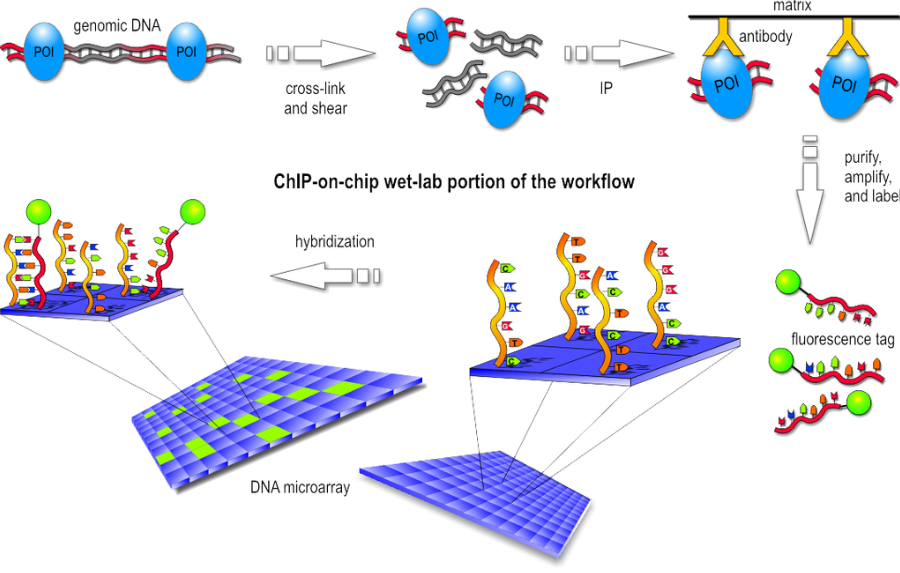
Basically the first step consist in the chromatin immunoprecipitation, like the one of ChIP-seq, but after the purification of DNA we have the labeling with a fluorescent probe. Finally, the fragments are poured over the surface of the DNA microarray, which is spotted with short, single-stranded sequences that cover the genomic portion of interest. Whenever a labeled fragment finds a complementary fragment on the array, they will hybridize and form again a double-stranded DNA fragment.
- Dry-lab step
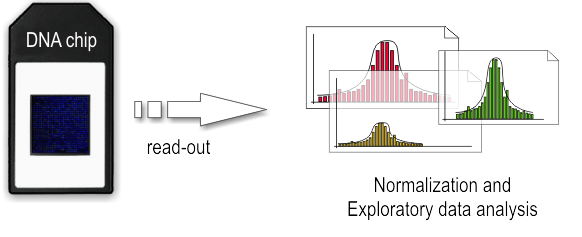
The array is illuminated with fluorescent light, the probes on the array that are hybridized to one of the labeled fragments emit a light signal that is captured by a camera. Than the captured fluorescence signals from the array are normalized. At this point it is possible to perform the analysis of the enriched regions and identify the binding sites.
c. ChIA-PET
Chromatin Interaction Analysis by Paired-End Tag Sequencing is a technique used to determine de novo long-range chromatin interactions genome-wide. In this method, DNA-protein complexes are crosslinked and fragmented. Specific antibodies are used to immunoprecipitate proteins of interest. Two sets of linkers, with unique barcodes, are ligated to the ends of the DNA fragments in separate aliquots, which then self-ligate based on proximity. The DNA aliquots are precipitated, digested with restriction enzymes, and sequenced. Deep sequencing provides base-pair resolution of the ligated fragments. So we can say that ChIA-PET can be used to identify unique, functional chromatin interactions between distal and proximal regulatory transcription-factor binding sites and the promoters of the genes they interact with.
ChIA-PET workflow:
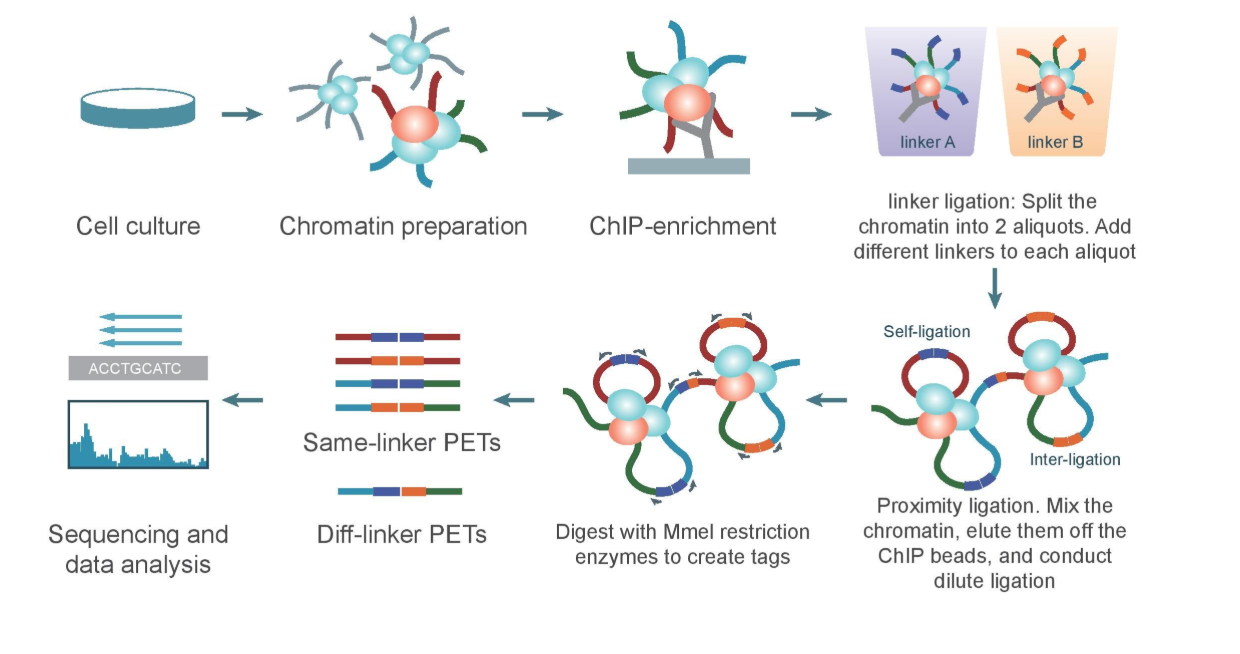
(Valeria Bastianini)
3. Biochemical determination of binding sequence
SELEX (Systematic Evolution of Ligands by Exponential Enrichment)
It was introduced in 1990.
Selex is a technique to determine the consensus-binding site of a TF without prior information.
The process begins with the synthesis of a very large oligonucleotide library consisting of randomly generated sequences of fixed length flanked by constant 5' and 3' ends that serve as primers (the sequence must be single strand). Library is incubated with immobilized target to allow oligonucleotide-target binding. Subsequently the sequences in the library are exposed to the target ligand (adapter). After the unbound oligonucleotides are washed away usually by affinity chromatography or target capture on paramagnetic beads. Then the bound sequences are eluted using denaturing solutions containing urea and EDTA or by applying high heat and physical force and amplified by PCR. These processes randomized single stranded library generation, incubation, binding, elution and amplification are repeated many time for the selection of sequences.

SELEX variants:
Instead of multiple rounds of binding and amplification, one round of selection at high stringency is sufficient, followed by elution and NGS sequencing.
- Hight-Throughput SELEX: the method utilizes massively parallel single-molecule sequencing technology, which eliminates all cloning steps and results in generation of a very large number of individual sequencing reads. The number of samples that can be analyzed in parallel is increased. The selected fragments can thus be directly sequenced without a ligation or template-switching step, decreasing the risk of sequence bias and DNA contamination. This method was developed for NGS.
- SELEX-Seq: differs from traditional SELEX in two respects the number of selected (bound) DNA oligos characterized (107 selected DNA oligos instead of 102) and the number of rounds of selection performed (one-two rounds)
Indirect technique that identifies regions where the TF binds but doesn't identify the motif sequence