Transcription Factor mapping and prediction
(Restore this version)
Modified: 4 April 2019, 4:45 PM User: Samar El Sherbiny →
Return to index
List of subjects:
1. TF binding site mapping genome-wide
2. TF sequence element prediction
3. biochemical determination of binding sequence (e.g. SELEX)
4. Activity assays for TFs
(Samar El Sherbiny)
TF binding site mapping genome-wide
The identification of transcription factor binding sites (TFBS) is an important initial step in determining the DNA signals that regulate transcription of the genome. We have different techniques able to identify this TFBS:
a. ChIP-seq
ChIP-seq is a technique that combines two aspects: chromatin immunoprecipitation with sequencing. It's a powerful method for identifying genome-wide DNA binding sites for transcription factors and other proteins. Following ChIP protocols, DNA-bound protein is immunoprecipitated using a specific antibody. The bound DNA is then coprecipitated, purified, and sequenced.
Advantages:
- We can analyze the entire genome;
- It reveals gene regulatory networks in combination with RNA sequencing and methylation analysis;
- It offers compatibility with various input DNA samples.
ChIP-seq workflow:
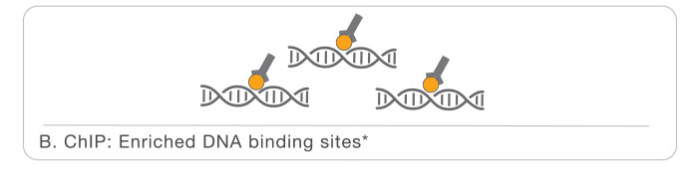
The first part is the Chromatin immunoprecipitation or library prep:
- Chromatin is crosslinked in order to fix all the interactions with proteins. Than it is fragmentated, for this step we can use different methods, but the most used is sonication.

- Than we have the enrichment, specific antibodies for the protein of interest are added to the sample in order to perform the immunoprecipitation.
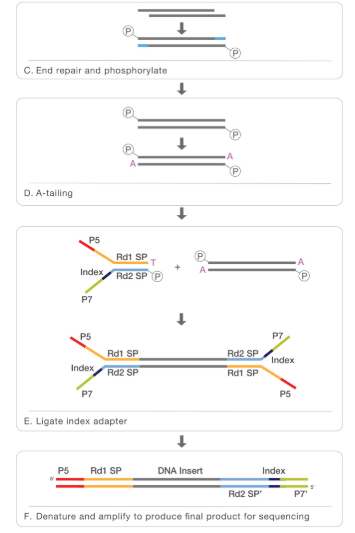
- At this point we can revert the crosslinking in order to obtain the DNA that will be purified. In order to perform NGS for the following step, that is sequencing, we will need also additional steps like end repair, phosphorilation, A-tailing, logation to the adapter and finally our sample will be ready for the sequencing.
The second part is the sequencing:
Before sequencing the fragments are amplyfied thanks to PCR and than we can have the sequencing, usually performed with NGS. In order to understand better all the steps of NGS sequencing i put a video from Illumina web site: https://www.youtube.com/watch?time_continue=290&v=fCd6B5HRaZ8
After sequencing the third part of ChIP-seq technique is the analysis of data:
Basically we obtain reads, they are small fragments that we can obtain after the sequencing. All the reads are mapped on the reference genome and we can see an enrichment in some spots: they are called peaks and from this output we can assume that the most enriched sites are with high probability the sites in which we have the binding between the transcription factor and the DNA. An important step in order to obtain data that are precise, is to estimate and eliminate the background. If we want to know the motif we need to perform the peak calling and peak annotation. Usually the binding site is around the center of the peak. To calculate the motif we need bioinformatic tools and specific algorithms.