Epigenomics: ChIP-Seq, DNase-Seq, FAIRE, ATAC-Seq, Nucleosome positioning
(Restore this version)
Modified: 10 June 2019, 5:12 PM User: Valery Sarlo →
[Audrey CHAMPIER]
ChIP-Seq
Methode used to analyse the interactions between proteins and DNA.
The technique consists in a chromatin immunoprecipitation followed by a parallel DNA sequencing in order to identify the binding sites of one protein on DNA. ( Before it, scientists used Chip on Chip : Chromatin immunoprecipitation + sequencing with DNA -microarray technique )
Chromatin immunoprecipitation : ( for more details see : " Wet-Lab portion of the workflow in the following link" : https://en.wikipedia.org/wiki/ChIP-on-chip#Workflow_of_a_ChIP-on-chip_experiment)
The protein of interest is first cross-linked with the DNA site it binds, thanks to formaldehyde for example.
Then the cells are lysed and the DNA , thanks to sonication, becomes small fragments.
After this , we introduce an antibody specific of the protein that interests us. This antibodies are bound to a surface and so we obtain,on this surface, all the sequence of DNA that bind the protein. In order to obtain only the DNA sequence, we reverse the cross-linking of DNA and proteins, thanks to heat.
Sequencing:
We use Parallel DNA sequencing method in order to obtain the sequence of all the fragments of DNA we just obtained with immunoprecipitation.
For more informations see:
https://en.wikipedia.org/wiki/ChIP-sequencing#ChIP
[Audrey CHAMPIER, Valery SARLO]
DNase-Seq
DNase seq is a method in molecular biology used to identify the location of regulatory regions.
For doing that, we use DNase-I, an enzyme able to cleave DNA sequences that are nucleosome-depleted so they are easier to cleave than the other sequences. This sequences are open chromatin and contains active regulatory regions.
So we fragment the DNA using DNase-I, we add biotinylated linker to extract and purify the DNA and we create a DNA library that will be sequencing (parallel DNA sequencing methods).
To sum up, we can say that DNase–seq experiments combine traditional DHS assays with high-throughput sequencing to simultaneously identify all types of regulatory regions genome-wide.
[Valentina Serra]
ATAC-seq
ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing) is a technology that can be used to obtain information about genome-wide chromatin accessibility. It is an emerging technique that is gaining popularity as it aids in a fast and sensitive analysis of the epigenome compared for example to the DNase-seq technology.
This method probes DNA accessibility using the hyperactive mutant Tn5 transposase which inserts sequencing adapters into accessible regions of the chromatin. The protocol is quite simple: after the lysis of the cells, the transposase complex fragments the genome and tags the resulting DNA with sequencing adapters. The tagged DNA fragments are then purified, amplified by PCR and sent for sequencing (using the linkers, already added by the enzyme, for the library preparation). More information can be found here: https://www.abcam.com/epigenetics/epigenetics-application-spotlight-atac-seq
The advantages of this technology are:
- the simplicity of the library preparation protocol
- very fast
- sonication or phenol-chloroform extraction are not required as for the FAIRE-seq; no antibodies like ChIP-seq; and no sensitive enzymatic digestion like MNase-seq or DNase-seq
The disadvantages are:
- during the mechanical sample processing, bound chromatin regions might open and be tagged by the transposase
- only half of the molecules contain the adapters in the orientation required for PCR amplification
[Isabella Tarulli]
FAIRE-Seq
FAIRE-Seq (Formaldehyde-Assisted Isolation of Regulatory Elements) is a method used for determining the sequences of DNA regions in the genome associated with a regulatory activity. In contrast to DNase-Seq, the FAIRE-Seq protocol doesn't require the permeabilization of cells or isolation of nuclei, and can analyze any cell type. FAIRE-Seq is based on differences in crosslinking efficiencies between DNA and nucleosomes or sequence-specific DNA-binding proteins. It uses the biochemical properties of protein-bound DNA to separate nucleosome-depleted regions in the genome. The protocol is based on the fact that the formaldehyde cross-linking is more efficient in nucleosome-bound DNA than it is in nucleosome-depleted regions of the genome. Cells are subjected to cross-linking, ensuring that the interaction between the nucleosomes and DNA are fixed. After sonication, the fragmented and fixed DNA is separated using a phenol-chloroform extraction. This method creates two phases: an organic and an aqueous phase. The DNA fragments cross-linked to nucleosomes will sit in the organic phase. Nucleosome depleted or open chromatin regions will be found in the aqueous phase. This assay then extracts the non cross-linked DNA and only these nucleosome-depleted regions will be purified, enriched and sequenced. FAIRE-extracted DNA fragments can be analyzed in a high- throughput way using next-generation sequencing. Sequencing provides information for regions of DNA not occupied by histones.
The advantages of this technology are:
- simple and highly reproducible protocol
- no antibodies required
- no enzymes required, such as DNase, avoiding extra steps necessary for enzymatic processing
- single-cell suspension or nuclear isolation not required, so the assay is easily adapted for use on tissue samples
The disadvantages are:
- No identification of regulatory proteins bound to DNA
- DNase-Seq may be better at identifying nucleosome-depleted promoters of highly expressed genes
Nucleosome positioning
We use the term “nucleosome positioning” to indicate where nucleosomes are located with respect to the genomic DNA sequence. The degree of positioning can vary from perfect positioning, in which a nucleosome is located at a given 147 bp stretch in all DNA molecules in a population, to no positioning, in which nucleosomes across a cell population are located at all possible genomic positions equally.
Nucleosome positioning is related to, but distinct from, the concept of nucleosome occupancy.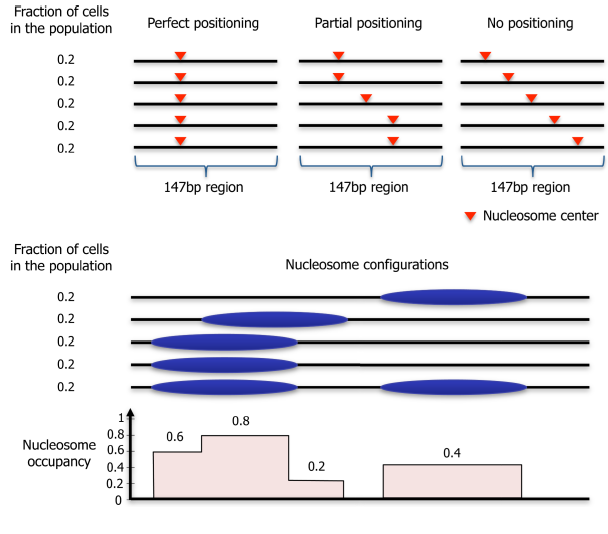
Nucleosome occupancy is the average number of nucleosomes measured within a specified genomic region in a cellular population and so is related to the probability of a nucleosome being present at the analysed site.
Nucleosome positioning is correlated to the probability of a reference point on a nucleosome existing at a specific genomic coordinate with respect to being present at the surrounding coordinates, in a given population of cells.
Nucleosome occupancy and positioning are critical to biological outcomes because nucleosomes inhibit the access of other DNA-binding proteins to the DNA.
DNA sequences can intrinsically favour or disfavour nucleosome formation, and have some intrinsic ability to influence the position of nucleosomes.
For optimal nucleosome formation, more bendable sequences are in contact with the histones, and less bendable sequences are solvent exposed. In particular, poly(dA:dT) and poly(dG:dC) tracts are intrinsically inhibitory to nucleosome formation, whereas non-homopolymeric GC-rich regions favour nucleosome formation. DNA sequence also contributes to the nucleosome positioning pattern.
[Martina De Luca]
Nucleosome positioning is an important chromatin feature that regulates gene expression. In particular, nucleosome depletion in the promoter and the precise positioning of the downstream nucleosomes play crucial roles in determining the transcription level, cell-to-cell variation, activation and repression dynamics, and might also function in defining the start and end points of transcribed regions. Nucleosomes affect transcription mostly by modulating the accessibility of regulatory factors and the transcriptional machinery to the underlying DNA sequence.
Nucleosome positioning across the genome is far from random and there is a difference between nucleosome density in regulatory regions respect to the transcribed ones. Indeed, in eukaryotes promoters are contained regions of DNA with very low nucleosome occupancy named nucleosome-depleted regions (NDRs), while sequences downstream of the transcription start site (TSS) are usually occupied by well-positioned nucleosomes.
Activators bind into NDRs, to specific region upstream the TSS, and induce the disassembling of neighbors nucleosomes and activate the gene. While repression is accompanied by chromatin reassembly, in most of the cases. In contrast to activators, Pol II assembly on promoters is more strictly anticorrelated with nucleosome occupancy. On the majority of the promoters, Pol II initiation sites are either exposed upon activation or located in constitutive NDRs. However, the initial Pol II binding is dictated by nucleosome positioning. Finally, the elongating polymerase has to go through a nucleosome barrier, so a large group of factors is involved in facilitating Pol II passage through the nucleosomes as well as maintaining the chromatin structure in this process.
The most common experimental approaches to identify nucleosome positions are
-the one which involves the use of restriction enzymes to evaluate them accessibility to DNA (if the site where the RE cuts is hampered by a nucleosome, it's unable to cut). This procedure is used in vitro experiments;
-the one that involves Micrococcal nuclease (MNase). MNase preferentially cuts linker DNA connecting two nucleosomes, while the nucleosomal DNA is at least partially protected against MNase digestion. After the MNase treatment the DNA is purified and treated with a RE. It's amplified and electrophoresis and Southern Blot are performed. Two are the possible results: if the nucleosomes are positioned 'as random', several bands appear; while if the nucleosomes are 'positioned nucleosomes' few well defined bands appear, that means that positioned nucleosome allow MNase cuts only at fixed points and nucleosomes are fixed in cells in the same site of the genome. This approach is used for in vivo experiments.
For more informations see:
- Bai, L., & Morozov, A. V. (2010). Gene regulation by nucleosome positioning. Trends in genetics, 26 (11), 476-483.
[ Anna Giulia Marcelli ]