Knocking-down and knocking-out genes

Written by ELISA QUARTA
Crispr-Cas9 technique
To Knock-down a gene means that its expression is reduced. To Knock-out a gene means that its expression is completely suppressed.
One way to generate a knockout mouse (so a mouse that completely lack for the expression of one gene) is using the technique CRISPR-Cas9.
CRISPR gene
editing is a genetic engineering technique in molecular biology by which the
genomes of living organisms may be modified. It is based on a simplified
version of the bacterial CRISPR-Cas9 antiviral defense system. By delivering
the Cas9 nuclease complexed with a synthetic guide RNA (gRNA) into a cell, the
cell's genome can be cut at a desired location, allowing existing genes to be
removed and/or new ones added in vivo.
Main components
CRISPR-Cas9 genome editing is carried out with a Type II CRISPR system. When utilized for genome editing, this system includes Cas9, crRNA, and tracrRNA along with an optional section of DNA repair template that is utilized in either non-homologous end joining (NHEJ) or homology directed repair (HDR). CRISPR-Cas9 often employs a plasmid to transfect the target cells, the main components of the plasmid are:
• crRNA --> Contains the guide RNA that locates the correct segment of host DNA along with a region that binds to tracrRNA (generally in a hairpin loop form), forming an active complex.
• tracrRNA --> Binds to crRNA and forms an active complex.
• sgRNA --> Single-guide RNAs are a combined RNA consisting of a tracrRNA and at least one crRNA.
• Cas9 --> An enzyme whose active form is able to modify DNA. Many variants exist with different functions (i.e. single-strand nicking, double-strand breaking, DNA binding) due to each enzyme's DNA site recognition function.
• Repair template --> DNA molecule used as a template in the host cell's DNA repair process, allowing insertion of a specific DNA sequence into the host segment broken by Cas9.
The crRNA is uniquely designed for each application, as this is the sequence that Cas9 uses to identify and directly bind to specific sequences within the host cell's DNA. The crRNA must bind only where editing is desired. The repair template is also uniquely designed for each application, as it must complement to some degree the DNA sequences on either side of the cut and also contain whatever sequence is desired for insertion into the host genome. Multiple crRNAs and the tracrRNA can be packaged together to form a single-guide RNA (sgRNA). This sgRNA can be included alongside the gene that codes for the Cas9 protein and made into a plasmid in order to be transfected into cells. Many online tools are available to aid in designing effective sgRNA sequences.
Structure
CRISPR-Cas9 offers a high degree of fidelity and relatively simple construction. It depends on two factors for its specificity: the target sequence and the PAM sequence. The target sequence is 20 bases long as part of each CRISPR locus in the crRNA array. A typical crRNA array has multiple unique target sequences. Cas9 proteins select the correct location on the host's genome by utilizing the sequence to bond with base pairs on the host DNA. The sequence is not part of the Cas9 protein and as a result is customizable and can be independently synthesized.
The PAM sequence on the host genome is recognized by Cas9. Cas9 cannot be easily modified to recognize a different PAM sequence. However, this is ultimately not too limiting, as it is typically a very short and nonspecific sequence that occurs frequently at many places throughout the genome (e.g. the SpCas9 PAM sequence is 5'-NGG-3' and in the human genome occurs roughly every 8 to 12 base pairs).
Once these sequences have been assembled into a plasmid and transfected into cells, the Cas9 protein with the help of the crRNA finds the correct sequence in the host cell's DNA and – depending on the Cas9 variant – creates a single- or double-stranded break at the appropriate location in the DNA.
Properly spaced single-stranded breaks in the host DNA can trigger homology directed repair, which is less error-prone than the non-homologous end joining that typically follows a double-stranded break. Providing a DNA repair template allows for the insertion of a specific DNA sequence at an exact location within the genome. The repair template should extend 40 to 90 base pairs beyond the Cas9-induced DNA break. The goal is for the cell's native HDR process to utilize the provided repair template and thereby incorporate the new sequence into the genome. Once incorporated, this new sequence is now part of the cell's genetic material and passes into its daughter cells.
Transfection
Delivery of Cas9, sgRNA, and associated complexes into cells can occur via viral and non-viral systems. Electroporation of DNA, RNA, or ribonucleocomplexes is a common technique, though it can result in harmful effects on the target cells. Chemical transfection techniques utilizing lipids have also been used to introduce sgRNAs in complex with Cas9 into cells. Types of cells that are more difficult to transfect (e.g. stem cells, neurons, and hematopoietic cells) require more efficient delivery systems, such as those based on lentivirus (LVs), adenovirus (AdV), and adeno-associated virus (AAV).
Double-strand breaks generation
The clustered regularly interspaced short palindrome repeats (CRISPR)/Cas9 system can be used to induce double-strand breaks (DSBs), single-strand nicks, or anywhere guide ribonucleic acids (RNAs) can bind with the protospacer adjacent motif sequence. By simple changing sequence of gRNA, Cas9-endonuclease can be delivered to a gene of interest and induce DSBs.
Utilizes in biomedicine
CRISPR-Cas technology has been proposed as a treatment for multiple human diseases, especially those with a genetic cause. Its ability to modify specific DNA sequences makes it a tool with potential to fix disease-causing mutations. Early research in animal models suggest that therapies based on CRISPR technology have potential to treat a wide range of diseases, including cancer, beta-thalassemia, sickle cell disease, hemophilia, cystic fibrosis, Duchenne's muscular dystrophy, Huntington's disease, and heart disease. CRISPR may also have applications in tissue engineering and regenerative medicine, such as by creating human blood vessels that lack expression of MHC class II proteins, which often cause transplant rejection.
I suggest this youtube video in order to understantd better this technology:
Please add other techniques to knockout or to knockdown a gene!
TALENS written by AKANDWANAHO Allan

Definition.
The abbreviation TALEN stands for Transcription Activator Like Effector Nucleases. They enable targeting of the genome with high precision. In 2011, this was named by Nature as the method of the year for gene editing. The Transcription Activator like Effectors (TALEs) were first discovered in the species of bacteria Xanthomonas genus and they were found to be secreted to the cytoplasm of plant cells infected by this bacteria from where they move into the nucleus. They are capable of DNA binding and activating the expression of their target genes by mimicking the eukaryotic transcription factors. They have two major domains; one for DNA binding and the other for cleavage (which is a nuclease). The DNA binding domain consists of monomers each binding to one nucleotide in the target sequence. For cleavage to occur, two TALENs must bind in close proximity to the target DNA, separated by a distance of 12-25bp. For more information please see http://www.genetherapynet.com/gene-editing-tools/talen.html and https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3547402/
Mechanism.
Binding of the DNA binding domains to a DNA double helix on specific sequences (one on the 5' end and the other on the 3' end) is followed by cleavage of the DNA sequence between the two TALENs by the nucleases attached to the DNA binding domains creating a double strand break between them. This maybe followed by cell repair mechanisms like NHEJ leaving deletions or insertions. This technique can therefore be used to knock out or knock in genes.

Please look at the following videos for further explanation
Applications
TALENs can be used just like the CRISPR/cas9 in genomic engineering https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4207558/. They can be used to generate knock out C. elegans, knock out mice and knock out zebra fish.
RNA interference
written by ASTRID SARACENI
RNA interference (RNAi) is a biological RNA-dependent gene silencing process (Knock-down) in which RNA molecules inhibit gene expression or translation, by neutralizing targeted mRNA molecules.
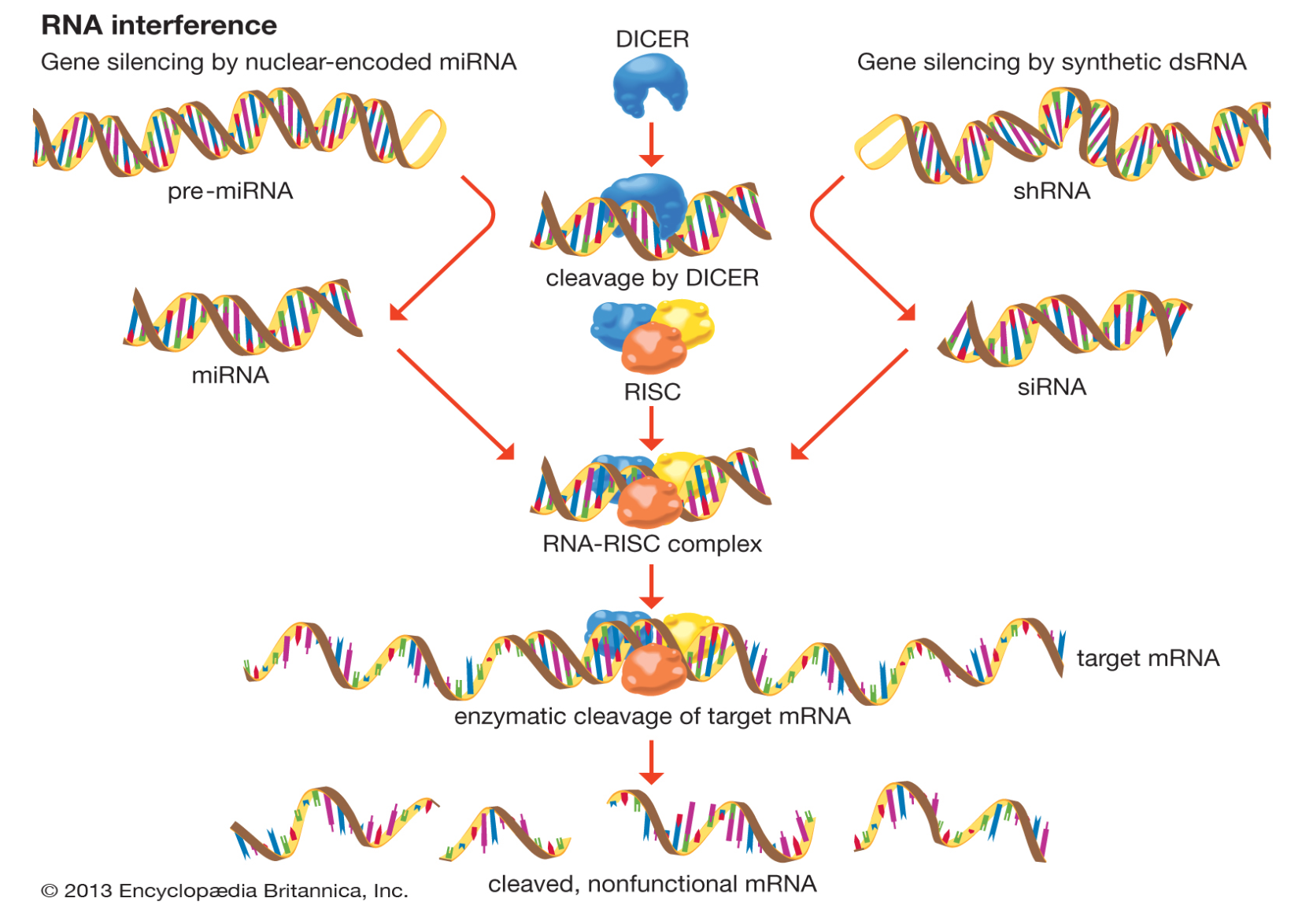
Two types of small RNA molecules – microRNA (miRNA) and small interfering RNA (siRNA) – are central to RNA interference. siRNAs derived from long dsRNA precursors differ from miRNAs in that miRNAs, especially those in animals, typically have incomplete base pairing to a target and inhibit the translation of many different mRNAs with similar sequences. In contrast, siRNAs typically base-pair perfectly and induce mRNA cleavage only in a single, specific target. RNAs are the direct products of genes, and these small RNAs can direct enzyme complexes to degrade messenger (mRNA) molecules and thus decrease their activity by preventing translation, via post-transcriptional gene silencing. Moreover, transcription can be inhibited via the pre-transcriptional silencing mechanism of RNA interference, through which an enzyme complex catalyzes DNA methylation at genomic positions complementary to complexed siRNA or miRNA.
The RNAi pathway is found in many eukaryotes, including animals, and is initiated by the enzyme Dicer, which cleaves long double-stranded RNA (dsRNA) molecules into short double-stranded fragments of ~21 nucleotide siRNAs. Each siRNA is unwound into two single-stranded RNAs (ssRNAs), the passenger strand and the guide strand. The passenger strand can be degraded, while the guide strand is incorporated into the RNA-induced silencing complex (RISC). The most well-studied outcome is post-transcriptional gene silencing, which occurs when the guide strand pairs with a complementary sequence in a messenger RNA molecule and induces cleavage by Argonaute 2 (Ago2), the catalytic component of the RISC. Small interfering RNAs can originate from inside the cell (as in pre-microRNAs expressed from RNA-coding genes in the genome) or can be exogenously introduced into the cell. When the dsRNA is exogenous (coming from infection by a virus with an RNA genome or laboratory manipulations), the RNA is imported directly into the cytoplasm and cleaved to short fragments by Dicer; while when it is endogenous, the primary transcripts from RNA-coding genes are first processed to form the characteristic stem-loop structure of pre-miRNA in the nucleus, then exported to the cytoplasm. Thus, the two dsRNA pathways, exogenous and endogenous, converge at the RISC.
RNAi is a valuable research tool, often exploited in experimental biology to study the function of genes both in cell cultures and living organisms, because synthetic dsRNA introduced into cells can selectively and robustly induce suppression of specific genes of interest.
Double-stranded RNA is indeed synthesized with a sequence complementary to a gene of interest and introduced into a cell or organism, where it is recognized as exogenous genetic material and activates the RNAi pathway. Using this mechanism, researchers can cause a drastic decrease in the expression of a targeted gene, and studying the effects of this decrease can show the physiological role of the gene product.
One problem of this tool could be the “off-targets”: when an introduced RNA has a base sequence that can pair with and thus reduce the expression of multiple genes. Such problems occur more frequently when the dsRNA contains repetitive sequences. A multitude of software tools have been developed implementing algorithms for the design of general mammal-specific, and virus-specific siRNAs that are automatically checked for possible cross-reactivity.
Specialized laboratory techniques have also been developed to improve the utility of RNAi in mammalian systems by avoiding the direct introduction of siRNA, for example, by stable transfection with a plasmid encoding the appropriate sequence from which siRNAs can be transcribed, or by more elaborate lentiviral vector systems allowing the inducible activation or deactivation of transcription, known as conditional RNAi.
 You can look this short video to better understand the RNAi mechanism: https://youtu.be/cK-OGB1_ELE
You can look this short video to better understand the RNAi mechanism: https://youtu.be/cK-OGB1_ELE