Transcriptome: special techniques, RNA-Seq, GRO-Seq, CAGE, others.
Back to index
RNA-seq
(Author: Ilaria Ferrarotto)
Transcriptome Analysis is the study of the transcriptome, of the complete set of RNA transcripts that are produced by the genome, under specific circumstances or in a specific cell, using high-throughput methods.
Transcriptome analysis by next-generation (RNA-seq) sequencing allows investigation of a transcriptome at unsurpassed resolution, detecting both coding and regulatory transcripts, like siRNA and lncRNA. One major benefit is that RNA-seq is independent of a priori knowledge on the sequence under investigation, thereby also allowing analysis of poorly characterized species.Brief outline of the workflow:
- bulk RNA is extracted from the sample and the desired RNA is selected (sample preparation)
- the selected RNA is copied into stable double-stranded copy DNA (library construction)
- the ds cDNA is then sequenced using various sequencing methods
- the sequences obtained can are aligned to reference genome sequences, available in data banks, to identify which genes are transcribed. This type of analysis provides a quantification of the expression levels for the transcribed genes. Alternatively, RNA-seq can be used to identify alternative splicing, novel transcripts, and fusion genes, following a new transcript discovery approach.


The complete workflow of RNA-seq consists of: (1) experimental design; (2) sample and library preparation; (3) sequencing; and (4) data analysis. You will find a general explanation of each step in the following video.
For a deeper understanding of the RNA-seq technology and its applications follow these links:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4648566/
GRO-Seq
Global Run-On sequencing is a high-throughput evolution of the Nuclear Run-On assay, introduced over 40 years ago, coupled to deep sequencing.
The advantage of this protocol is the exceptional sensitivity and the possibility to map nascent transcripts at the genome-wide scale providing a reliable and unbiased, real-time measure of transcriptional activity from engaged RNA polymerase in mammalian cells; in fact the steady-state level of RNA, measured by conventional sequencing methods, does not accurately mirror transcriptional activity per se.
Moreover it delivers a high-resolution map of coding and noncoding transcripts that is especially useful for annotation and quantification of short-lived RNA molecules, usually hard to detect because, owing to their instability, these transcripts do not accumulate in the nucleus and elude most RNA detection protocols.
For example, with this method it has been recently characterized enhancer-associated RNAs (eRNAs) and their transcription in response to stimuli such as estrogen, LPS and Epidermal Growth Factor; we have achieved crucial information on RNA polymerase II (RNAPII) such as density at different classes of protein coding genes, defects in elongation, pause-release and termination and the capacity to fire bi-directionally at most mammalian promoters, initiating noncoding RNAs that are transcribed antisense with respect to the messenger RNA.
Limitations: laboriousness of the technique and the amount of starting material (the number of cells that are required lies in the 10ˆ7 range)
Protocol:
- Nuclei isolation: Nuclei from mammalian cells are isolated, washed to remove free nucleotides and kept at ice-cold temperature to arrest ongoing transcription;
Nuclear Run-On: Transcription is resumed in vitro when nuclei are incubated at 30°C in the presence of brominated nucleotides and the anionic detergent sarkosyl, which prevents de novo assembly of the pre-initiation complex and avoids re-initiation;
Elongation: Transcripts that were initiated at the time of nuclei isolation (commonly referred to as nascent RNA ) will be further elongated by engaged RNA polymerase, to allow incorporation;
Firts immunoprecipitation: affinity purification by means of commonly used antibodies against bromodeoxyuridine (anti-BrdU);
End repair;
Second immunoprecipitation;
Adapter ligation;
Third immunoprecipitation;
Library preparaton: isolate nascent RNA can be ultimately converted into a Illumina-compatible DNA library suitable for deep sequencing;

Sources:
- Gardini A. (2017) Global Run-On Sequencing (GRO-Seq). In: Ørom U. (eds) Enhancer RNAs. Methods in Molecular Biology, vol 1468. Humana Press, New York, NY, DOI: https://doi.org/10.1007/978-1-4939-4035-6_9
- GRO-seq, A Tool for Identification of Transcripts Regulating Gene Expression, March 2017, Methods in Molecular Biology 1543:45-55, DOI: 10.1007/978-1-4939-6716-2_3
CAGE -seq
The begging
Moving from Sanger to next-generation sequencing, the refinement of CAGE technology has gone alongside the development of sequencing technology, which clearly gave us the power to characterize RNA better than before.
Introduction
CAGE stands for Cap-Analysis gene expression, that means it analyzes 5' cap of mRNA, but not only, it helps to identify and quantify the transcriptional start sites (TSSs), within promoters are characterized at single nucleotide resolution. CAGE allows to map of all the initiation sites of both capped coding and noncoding RNAs. Even to identify novel regulatory elements, the predictions of transcription factor binding sites and motifs associated with transcription.
The analysis of 5’ ends by CAGE, in eukaryotes, it is suitable to imply gene regulatory networks and it has provided knowledge of the key transcription factors responsible for the differentiation of cell, for instance of monoblasts to monocytes (Suzuki H, et al.).
Deeper sequencing is necessary to detect all active promoters in a given tissue, for instance in mammalian cells, since they have at least 5–10 time more TSS. CAGE was used to discover promoter activity from small subpopulations of hippocampal cells (Valen, Eivind (2009).
ENCODE project at NIH is one of the most important database that use this technique.
The picture below show us the general workflow of CAGE.

Also CAGE allow the operator to observe that retrotransposon elements are specifically expressed and act as regulators of protein coding RNAs and other ncRNAs.
How it works
The CAGE utilizes a “cap-trapping” technology based on the biotinylation of the 7-methylguanosine cap of Pol II transcripts, to pull down the 5’-complete cDNAs reversely transcribed from the captured transcripts. Through a massive sequencing of the 5’ end of cDNA and analysis of the sequenced tags, transcription start sites and transcripts amount are inferred on a genome-wide scale.
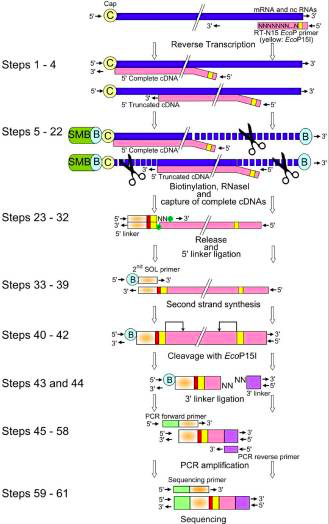
The main steps of CAGE are:
- reverse transcription with random primer mixture to make cDNA
- Biotinylation: biotin hydrazide, generated by oxidation process
- ssRNA digestion with RNAse1
- Capture of the fragments by magnetic beads
- wash away
- Released cDNA from mRNA by denaturation
- single strand linker ligation in which the raptor carries barcode at the 3' end of cDNA
- Single strand linker ligation at 5' end cDNA
- 2nd stand synthesis by longer linker primer
- loaded on the instrument and sequenced
Analyzing data
The primary output of CAGE is a set of sequences, each of which represents a short reads corresponding to the 5’ end of capped RNA molecules, also called CAGE tags. after that will follow the computational processing from which we can obtain a mapping, so genomic location, clustering aggregation into a unit of transcriptional initiation on genome and tags activity or expression level.
Pros
- Measures RNA expression levels
- Maps TSS in promoter regions at single-nucleotide resolution
- Discover alternative promoters
Limitations
- Only works on total mature RNA
- CAGE selectively removes non-capped RNAs
- CAGE is not applicable to prokaryotes or RNAs shorter to 100 nt
(written by Dante Davide)
sources:
Hazuki Takahashi, Timo Lassmann, Mitsuyoshi Murata, and Piero Carninci, 5’ end-centered expression profiling using Cap-analysis gene expression (CAGE) and next-generation sequencing, 2012, Nature Protocol, 542- 561
Valen and Eivind, Genome-wide detection and analysis of hippocampus core promoters using DeepCAGE, 2009, Genome Research, 255–265.
Rimantas Kodzius et al., CAGE: cap analysis of gene expression, Nature Methods, 2006, 211–222.
name="toc-10">SAGE seq
The Begging
Introduction
- A small sequence of nucleotides from the transcript, called a ‘tag’, can effectively identify the original transcript from whence it came.
- Linking these tags allows for rapid sequencing analysis of multiple transcripts.
How SAGE works
Few steps for SAGE
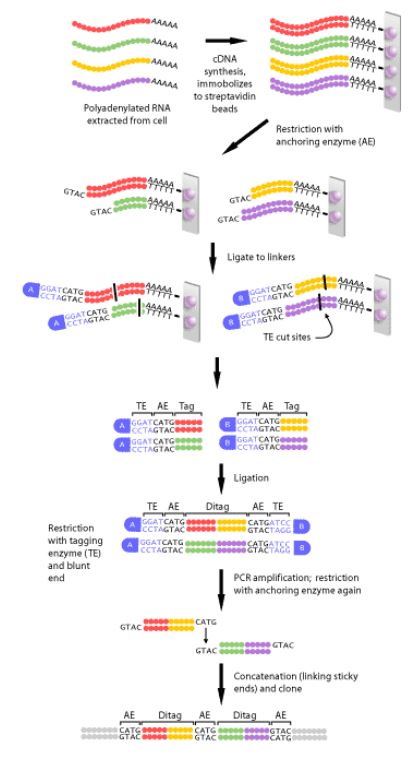
- Isolate RNA: DNA extraction is a critical first step, a complimentary DNA strand, or cDNA, of each transcript in the cell must be generated. This is necessary, since mRNA is much less stable than DNA.
- Perform PCR
- Perform sequencing reaction
- Purify the sequencing reaction: it is important to remove unincorporated dye terminators and salts that may compete
- Perform capilalry electrophoresis
- Analyze data
Pro
- mRNA sequence doesn't need to be known prior, so genes of variants whihc are not known can be discovered.
- its more accurate as it involves direct counting of the number of transcript.
Limitations
- teh length of geen tag is extremly short, about 13 bp, so if the tag is derived form and unknown gene, it's difficult to analyze with such a short sequence.
- Type 2 restriction enyme doesn't yield same length fragments.
- mRNA levels and protein expresison are not always correlated.